
Résumé
Il est classique de faire remonter à James Lind en 1747 l’histoire du premier essai clinique dans la prévention du scorbut et à Claude Bernard en 1865 l’établissement des bases de la médecine expérimentale. Cependant il faut attendre l’essor des thérapeutiques au milieu du 20e siècle et l’évolution des concepts en méthodologie pour que soit réalisé le premier essai clinique comparatif randomisé en 1948 montrant les effets de la streptomycine significativement différents du placebo dans le traitement de la tuberculose pulmonaire. La ‘‘ médecine factuelle ’’ d’aujourd’hui, ou ‘‘ evidence-based medicine ’’ des anglo-saxons, correspond à un souci de rationalisation des choix thérapeutiques. Elle repose sur les résultats des essais cliniques contrôlés qui garantissent le niveau de preuve le moins discutable et le plus fiable. Pour être valable, un essai clinique contrôlé doit remplir des conditions méthodologiques précises. Il doit avoir un objectif principal qui est de confirmer ou d’infirmer une hypothèse préalable. Le choix du critère principal de mesure d’effet doit découler de l’objectif de l’étude. L’essai doit comparer les résultats dans un groupe de patients traités par le médicament à tester à ceux observés dans un groupe de patients témoins, le groupe ‘‘ contrôle ’’, les deux groupes étant suivis simultanément. L’attribution des traitements aux patients doit être faite par tirage au sort et l’observation des patients ainsi que l’analyse des résultats doivent être réalisées en aveugle des traitements administrés. Un calcul préalable du nombre de patients à inclure dans l’essai doit être effectué en tenant compte du type d’essai choisi (supériorité ou équivalence), de l’hypothèse que l’on veut confirmer, des risques statistiques et de la variabilité du critère principal de mesure d’effet. Le plan expérimental de l’essai, croisé ou en groupes parallèles, doit être adapté à l’objectif principal de l’étude et aux caractéristiques de la pathologie. Enfin, l’analyse des résultats de l’essai doit être effectuée en intention de traiter, c’est à dire en tenant compte de tous les patients inclus et randomisés dans l’essai. Si les résultats de ces essais méthodologiquement corrects servent effectivement à élaborer des recommandations thérapeutiques officielles pour les médecins, il n’en est pas moins vrai que certaines limitations doivent être prises en compte lorsqu’on veut transposer les résultats expérimentaux dans la pratique médicale usuelle. Les patients sélectionnés pour les essais cliniques, ne représentent pas forcément la totalité de la population des patients qui seront exposés au traitement par la suite. La durée des traitements dans les essais est souvent très inférieure à celle des traitements dans la pratique médicale. Par ailleurs, le nombre des patients inclus dans les essais est forcément limité ce qui diminue la capacité des essais à détecter des effets indésirables rares. Enfin, la prise en charge des malades dans les essais cliniques est différente de celle de la pratique médicale usuelle. Ces limites justifient que soient réalisées des études pharmaco-épidémiologiques de cohortes de patients exposés au médicament dans ‘‘ la vraie vie ’’ lorsqu’on estime insuffisantes les informations sur le médicament collectées dans les essais cliniques contrôlés, avant l’AMM. Malheureusement, il existe des domaines de la recherche biomédicale qui souffrent de l’absence d’essais cliniques contrôlés soit parce qu’ils n’intéressent pas les promoteurs industriels des médicaments soit parce qu’ils concernent des problèmes de santé publique qui n’ont pas à être résolus par des investisseurs privés. Dans ces cas, seule une recherche médicale soutenue par des investissements publics institutionnels permet de réaliser des essais cliniques contrôlés. Ce sont aujourd’hui les organismes publics nordaméricains qui assurent ces investissements et il est très dommage que l’Europe ne joue pas un rôle plus important dans ce domaine. Le progrès des connaissances médicales dans le diagnostic et le traitement des maladies repose aujourd’hui essentiellement sur les résultats des essais cliniques contrôlés. Encore faut-il que la population de patients inclus dans ces essais soit similaire à la population française afin que les résultats des essais lui soit transposable ! Ce problème important ne sera résolu que lorsque de grands essais cliniques contrôlés seront organisés en Europe afin d’apporter aux médecins européens les résultats probants dont ils ont besoin pour conforter leur pratique.
Summary
It is generally agreed that the first comparative clinical trial in history was done by James Lind in 1747, in the treatment of scurvy. The general bases of modern experimental medicine were published by Claude Bernard in 1865. However, it is the development of new drugs and the evolution of methodological concepts that led to the first randomized controlled clinical trial, in 1948, which showed that the effects of streptomycin on pulmonary tuberculosis were significantly different from those of a placebo. Today, ‘‘ evidence-based ’’ medicine aims to rationalize the medical decision-making process by taking into account, first and foremost, the results of controlled randomized clinical trials, which provide the highest level of evidence. In the second half of the 20th century it became clear that different kinds of clinical trials might not provide the same level of evidence. Practitioners’ intimate convictions must be challenged by the results of controlled clinical trials. Take the CAST trial for example, which, in 1989, tested antiarrhythmic drugs versus placebo in patients with myocardial infarction. It was well known that ventricular arrhythmias were a factor of poor prognosis in coronary heart disease, and it was therefore considered self-evident that drug suppression of these ventricular arrhythmias would reduce the mortality rate. In the event, the CAST trial showed the exact opposite, with an almost 3-fold increase in total mortality among patients with coronary heart disease who were treated with antiarrhythmic drugs. These results had a profound impact on the use of antiarrythmic drugs, which became contraindicated after myocardial infarction. A clinical trial has to fulfill certain methodological standards to be accepted as evidence-based medicine. First, a working hypothesis has to be formulated, and then the primary outcome measure must be chosen before beginning the study. An appropriate major endpoint for efficacy must be selected, in keeping with the primary outcome. One may choose either a single endpoint (for instance all-cause mortality) or a composite criterion taking into account various manifestations of the same health disorder (for instance cardiovascular mortality plus non lethal myocardial infarction plus non lethal ischemic stroke). The trial must be controlled, i.e. must compare the intervention with a standard or dummy treatment. A randomization process is used to ensure that the groups are comparable. The patients must be monitored and the results analyzed in double-blind manner. The required number of patients is calculated based on the working hypothesis (‘‘ superiority ’’ trial or ‘‘ equivalence ’’ trial), as well as the spontaneous variability of the main endpoint, and the alpha and beta statistical risks. The experimental design (cross-over or parallel groups) is chosen according to the primary outcome measure and the disease characteristics. Finally, the results must be analyzed in an intention-to-treat manner, taking into account all the patients who were initially randomized. The results of these methodologically sound trials form the basis for official therapeutic guidelines, which help physicians to choose the best treatments for their patients. However, extrapolating the results of randomized controlled clinical trials to the general patient population is not always straightforward. For instance, it is well known that patients who participate in clinical trials are highly selected and therefore somewhat unrepresentative. In addition, their numbers are limited and the treatment period is often much shorter than in routine management of a chronic disease. Finally, patients in clinical trials are monitored more closely than in routine practice. This is why we need post-marketing pharmacoepidemiological studies, in which cohorts of patients exposed to the treatment in question are monitored sufficiently long to determine the precise risk-benefit ratio. Controlled clinical trials are lacking in various fields of biomedical research, either because drug companies consider them unprofitable, or because they concern public health issues that are outside the scope of the private sector. In such cases, controlled clinical trials must be undertaken and funded by the public sector. Today, only North American institutions such as NIH and the National Heart Blood and Lung Institute are capable of sponsoring such trials. This creates a potential problem for extrapolation to European patient populations, which may be different. Large controlled clinical trials must start to be sponsored by public funding in Europe if European practitioners are to receive the evidence-based results they need to rationalize their medical practice.
INTRODUCTION
Il est classique d’attribuer à James Lind la paternité du premier essai clinique contrôlé publié en 1753 dans son livre ‘‘ A treatise of the scurvy ’’ [1]. James Lind, né en 1716, était médecin de marine lorsqu’il réalisa en 1747 à bord du Salisbury, un navire de la Royal Navy, l’expérience suivante qui visait à tester différents remèdes contre le scorbut. Le scorbut décimait les équipages et, comme l’écrit Lind, ‘‘ avait tué plus de marins anglais durant la dernière guerre que les armes réunies de la France et de l’Espagne ’’. Après avoir sélectionné douze marins atteints de lésions scorbutiques, il les répartit en six groupes de deux afin de comparer entre les groupes les effets de l’acide sulfurique dilué, de l’eau de mer, d’un mélange de moutarde, ail et raifort, du cidre, du vinaigre et dans le dernier groupe, de deux oranges et un citron chaque jour. Après quinze jours de traitement, ‘‘ les deux qui firent usage des oranges et des citrons reçurent le soulagement le plus prompt et le plus sensible ’’.
Lind venait de réaliser le premier essai clinique comparatif en ouvert, sur six groupes parallèles de patients. L’essai n’était probablement pas randomisé car Lind ne précise pas si l’attribution de chaque traitement aux douze hommes avait été réalisée par tirage au sort ou non. Quoiqu’il en soit, la conclusion s’imposait :
oranges et citrons guérissaient les lésions scorbutiques alors que les autres remèdes étaient sans effet. Malheureusement, Lind ne sut pas développer sa découverte [2].
Employant ensuite un concentré de jus de citron en bouteille, il ne parvint pas à renouveler l’expérience et nous savons pourquoi : la vitamine C, rapidement oxydée à l’air libre, n’était active dans la première expérience que parce qu’il s’agissait de jus de fruits frais. James Lind publia son traité sur le scorbut en 1753, fut élu membre du Royal Collège des Médecins puis de l’Académie Royale de Médecine de Paris en 1776 avant de s’éteindre en 1794. Il fallut attendre quarante ans après l’expérience initiale pour que des citrons soient distribués aux équipages anglais, et cent ans après l’expérience pour que le scorbut cessât de décimer les équipages français.
Il nous faut donc considérer l’essai clinique de James Lind comme l’essai fondateur de l’expérimentation clinique, plus d’un siècle avant que Claude Bernard publie au Collège de France en 1865 sa célèbre ‘‘ Introduction à l’étude de la médecine expérimentale ’’ qui allait poser les bases du raisonnement expérimental dans lequel l’hypothèse doit être confirmée ou infirmée par l’expérience clinique en comparant des groupes de patients traités simultanément par différentes méthodes. Pour la première fois dans l’histoire de la médecine, l’expérimentation clinique de Lind comparait des traitements administrés simultanément à des groupes de patients et arrivait à ‘‘ une déduction empirique là où la théorie était insuffisante pour expliquer les observations ’’ [2]. Il faut citer ensuite le travail de pionnier que publia Pierre Louis en 1835 montrant dans un essai contrôlé l’inefficacité clinique de la saignée dans le traitement des maladie humaines telles que la pneumonie, l’érysipèle et l’angine [3]. Mais il fallut attendre 1948 pour voir publier les résultats du premier essai clinique contrôlé (versus placebo) randomisé qui démontra les effets bénéfiques de la streptomycine dans le traitement de la tuberculose pulmonaire [4]. Les fondements de l’expérimentation clinique étaient posés, comme le résume JeanPierre Boissel [5] :
— l’hypothèse à tester est formulée avant que soient collectés les résultats de l’expérience — un raisonnement statistique est utilisé pour tenir compte de la variabilité de la maladie, des effets du traitement et des caractéristiques des patients — la preuve de la relation causale est fondée sur la comparaison entre un groupe de patients traités et un groupe témoin de patients comparables, la répartition des patients entre ces groupes étant réalisée par tirage au sort.
LA MÉDECINE FACTUELLE
La médecine expérimentale s’est imposée aujourd’hui dans le raisonnement médical et la pratique médicale fait référence de nos jours de plus en plus à la ‘‘ médecine factuelle ’’, ‘‘ l’Evidence-Based Medicine ’’ des anglo-saxons. Sa définition proposée par Gilles Bouvenot est la suivante : ‘‘ médecine dont les pratiques sont directement inspirées de faits scientifiques validés d’interprétation indiscutable ’’ [6]. Cette approche, développée dans les années 1990 à l’Université McMaster d’Ontario au Canada par un groupe de chercheurs auquel participait un français, notre collègue Joël Ménard, est devenue le fondement du raisonnement médical [7]. Devant un problème médical diagnostique ou thérapeutique, le médecin doit d’abord chercher dans la littérature médicale les informations qui lui sont nécessaires pour éclairer sa décision. Chaque information doit être évaluée en fonction du niveau de preuve des résultats qu’elle présente, c’est à dire qu’elle doit être passée au crible d’une analyse critique dans le but de déterminer si ces résultats ont été obtenus selon une méthodologie correcte et si leur authenticité et le niveau de preuve qu’ils apportent les rend entièrement acceptables par le médecin.
Dans la classification actuelle des niveaux de preuve fournis par la littérature médicale (tableau 1), l’essai clinique contrôlé randomisé apporte les résultats les plus fiables et les plus robustes (niveau de preuve 1A) qui peuvent être utilisés par les médecins pour rationaliser leur démarche diagnostique et thérapeutique [8].
L’objectif de ce travail est de montrer pourquoi les essais cliniques contrôlés randomisés constituent l’élément primordial de la médecine factuelle et d’en analyser les caractéristiques méthodologiques essentielles ainsi que les limites opérationnelles.
UN EXEMPLE ILLUSTRANT L’INTÉRÊT DES ESSAIS CLINIQUES CONTRÔLÉS RANDOMISÉS
L’intérêt des essais cliniques contrôlés randomisés est de soumettre à une démarche expérimentale ‘‘ l’intime conviction ’’ du médecin prescripteur, c’est à dire de l’amener à fonder sa pratique sur la prise en compte de résultats expérimentaux objectifs plutôt que sur des arguments subjectifs reposant sur des expériences individuelles non contrôlées, ou sur des impressions non validement étayées, ou sur des prises de position présentées comme certaines par des leaders d’opinion mais qui ne résistent pas à une analyse critique objective. L’exemple des médicaments anti-arythmiques dans les années 1980 illustre la nécessité de cette démarche. Depuis l’utilisation de l’électrocardiogramme (ECG) et surtout de son enregistrement sur bande magnétique pendant vingt-quatre heures selon la méthode de Holter, les cardiologues savaient que la présence d’extrasystoles ventriculaires (ESV) était un facteur de mauvais pronostic chez des patients dans les suites d’un infarctus du myocarde. De
TABLEAU no 1. — Classification des niveaux de preuves (d’après COOK D.J. et al. ) [8]
Niveau de preuve Méthodes d’étude aboutissant à des résultats 1A Grands essais cliniques comparatifs 1 contrôlés et randomisés 1B Méta-analyses méthodologiquement correctes 2 Petits essais comparatifs randomisés Grands essais randomisés dont les résultats demeurent incertains 3 Essais comparatifs non randomisés, avec un groupe contrôle contemporain Suivis de cohortes de patients 4 Essais comparatifs non randomisés, rétrospectifs ou avec un groupe contrôle historique Etudes cas-témoins 5 Essais sans groupe contrôle Séries de malades Essais réalisés sur des critères intermé- diaires non substitutifs Consensus professionnels ou opinions d’experts nombreux travaux [9-12] avaient montré que la présence de ces ESV permettait d’identifier des patients à haut risque de mort subite après l’infarctus. La grande fréquence et la variabilité de forme de ces ESV constituaient autant de facteurs pronostiques aggravants. Mukahrji et al. [12] avaient même montré que les patients ayant plus de dix ESV par heure d’enregistrement de l’ECG présentaient un risque de décès quatre fois supérieur à celui des patients ayant moins de dix ESV par heure, et ceci même après ajustement sur la fraction d’éjection ventriculaire gauche de ces patients.
Puisqu’on disposait depuis les années 1970 de médicaments anti-arythmiques puissants capables de diminuer le nombre des ESV — voire même de les supprimer, « l’intime conviction » des cardiologues était donc de considérer comme établi le fait que la suppression de ces ESV par les anti-arythmiques permettait de diminuer le risque de mort subite après un infarctus du myocarde et d’améliorer le pronostic vital de ces patients. Considérant le résultat comme allant de soi, les cardiologues élaboraient des stratégies thérapeutiques complexes et les laboratoires pharmaceutiques participaient à la découverte de nouveaux médicaments anti-arythmiques de plus en plus puissants capables d’éradiquer l’ECG de ces ESV de mauvais augure.
Il fallut attendre 1988, pour que le National Heart Lung and Blood Institute américain entreprenne une première étude pilote, CAPS ou Cardiac Arrythmia
Pilot Study, afin de valider cette stratégie et tester l’hypothèse selon laquelle la suppression des arythmies ventriculaires par les anti-arythmiques après un infarctus du myocarde pouvait améliorer la survie des patients [13]. Cette première étude de faisabilité fut menée chez 507 patients ayant plus de dix ESV par heure sur un Holter de 24h, et une fraction d’éjection ventriculaire gauche supérieure à 20 %, inclus dans l’étude 6 à 60 jours après un infarctus du myocarde. Cet essai clinique, réalisé en cinq groupes parallèles randomisés, permit de tester en double-aveugle plusieurs antiarythmiques en augmentant les doses ou en changeant d’anti-arythmique jusqu’à ce qu’on aboutisse à une diminution d’au moins 70 % de la fréquence des ESV et d’au moins 90 % des salves d’ESV. A la suite de cet essai, trois médicaments efficaces et bien tolérés furent sélectionnés l’encainide, le flecainide et la moricizine. Le NHLB Institute entreprit alors une deuxième étude en double aveugle avec ces trois antiarythmiques, l’étude CAST (Cardiac Arrythmia Suppression Trial) [14]. Sur 2 309 patients pré-selectionnés, 1 727, ayant les mêmes caractéristiques que ceux de la première étude CAPS et la même réponse aux traitements anti-arythmiques, furent progressivement inclus dans l’étude et randomisés en quatre groupes parallèles (un groupe encainide, un groupe flecainide, un groupe morizicine et un groupe placebo).
Après un suivi de dix mois, le comité de suivi de l’étude CAST recommanda le 16 Avril 1989 d’arrêter l’étude chez les patients traités par encainide ou flecainide car la mortalité y était significativement supérieure à celle du placebo. L’évolution de la survie dans ces deux groupes par rapport au placebo est illustrée par les figures no 1 (mortalité due à une arythmie ou à un arrêt cardiaque) et no 2 (mortalité toutes causes confondues). On comptait 56 décès sur 730 patients (7,7 %) dans les groupes des anti-arythmiques contre 22 décès sur 725 patients (3 %) dans le groupe placebo.
Ces premiers résultats publiés le 10 Août 1989 [14] furent suivis d’une publication complète deux ans plus tard [15] qui confirmait une mortalité de 8,3 % dans les groupes anti-arythmiques contre 3,5 % dans le groupe placebo, différence significative avec p. = 0,0001.
Ainsi donc, un essai clinique de grande envergure, randomisé, contrôlé versus placebo réalisé en double aveugle, venait clairement d’infirmer l’hypothèse initiale et de montrer que, contrairement à ce que pensait la majorité des cardiologues, le traitement anti-arythmique après un infarctus du myocarde était délétère pour les patients, même si les ESV avaient considérablement diminué sous traitement. Les anti-arythmiques normalisaient l’ECG mais augmentaient la mortalité des patients.
Le résultat de l’étude CAST dont on voit l’impact majeur sur la place des médicaments anti-arythmiques dans le traitement des patients après un infarctus du myocarde, furent l’objet de très nombreuses controverses. Les résultats étaient difficiles à accepter mais ils étaient là et la pratique médicale en fut profondément modifiée. La prescription des anti-arythmiques en fut considérablement restreinte et notamment contre-indiquée chez les patients souffrant d’une ischémie myocardique. Cet exemple montre bien comment un essai clinique méthodologiquement correct avait abouti à modifier l’attitude thérapeutique des cardiologues.
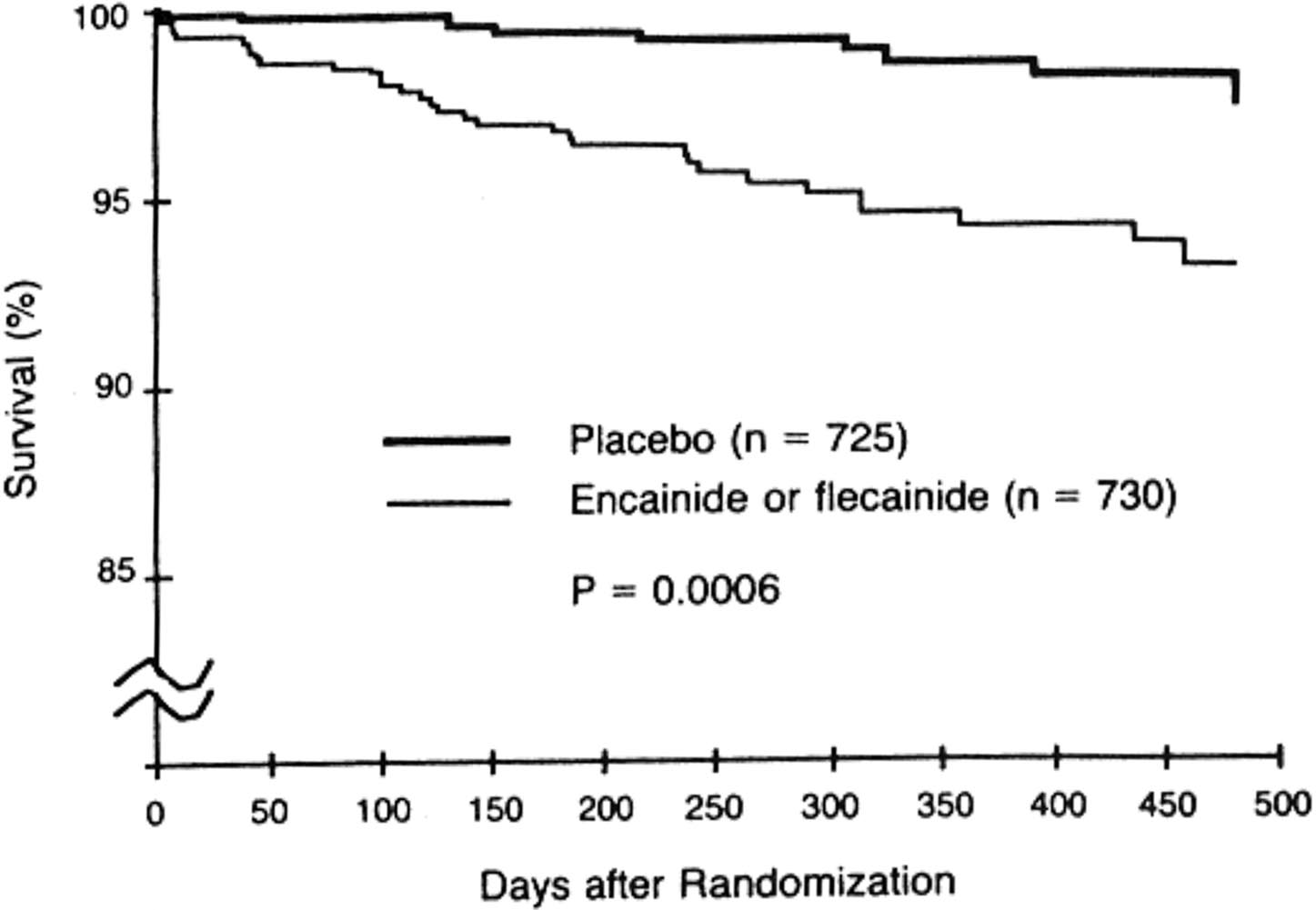

TABLEAU no 2 : Les principales caractéristiques méthodologiques des essais cliniques contrôlés — L’essai doit être comparatif et ‘‘ contrôlé ’’, c’est-à-dire doit comporter un groupe témoin de patients traités simultanément — L’attribution des traitements aux patients doit être réalisée par tirage au sort (randomisation) — Le suivi des patients et l’analyse des résultats doivent être réalisés en aveugle des traitements administrés — Les critères de mesure d’effet doivent être définis a priori et différencier le critère principal des critères secondaires — Le calcul du nombre de patients à inclure dans l’essai clinique contrôlé obéit à des règles précises — Le plan expérimental doit être adapté à l’objectif principal de l’essai — L’analyse des résultats doit être réalisée en intention de traiter LES CARACTÉRISTIQUES METHODOLOGIQUES DES ESSAIS CLINIQUES CONTRO
Ù
LÉS RANDOMISÉS
Pour être valable, l’essai clinique contrôlé doit obéir à des règles méthodologiques précises.
Les sept caractéristiques méthodologiques principales des essais cliniques contrôlés randomisés sont aujourd’hui parfaitement établies [6] (tableau no 2).
— L’essai doit être contrôlé. Ceci signifie qu’il s’agit d’un essai comparatif permettant de comparer simultanément les effets d’un traitement dans un groupe traité, par exemple par un nouveau médicament, à ceux d’un traitement de référence dans un groupe contrôle, traitement témoin qui peut être un médicament déjà connu ou un placebo. (Notons au passage que, par anglicisme, le groupe ‘‘ témoin ’’ est devenu le groupe ‘‘ contrôle ’’ dans la totalité des publications françaises sur le sujet !). Le choix du placebo ou d’un ‘‘ médicament de réfé- rence ’’ dans le groupe témoin est un problème complexe car l’utilisation d’un placebo en monothérapie n’est possible que si elle n’entraîne pas de perte de chance thérapeutique pour les patients qui y sont soumis. La question est donc d’ordre éthique. Dans les pathologies graves où le pronostic vital est en jeu et où existent déjà des médicaments qui ont fait la preuve d’un vrai bénéfice thérapeutique pour les patients (diminution de morbidité et/ou de mortalité), il n’est pas possible de priver les patients de ces traitements déjà validés. L’exemple de cette situation est apporté par l’étude CIBIS II [16] où les patients en insuffisance cardiaque de classe NYHA 3 ou 4 ne pouvaient pas être privés de diurétiques et d’inhibiteurs de l’enzyme de conversion (IEC) de l’angiotensine II, qui avaient
prouvé leurs effets sur la diminution de la mortalité. Dans ce cas, le médicament de l’essai, un bêta-bloquant le Bisoprolol, a été attribué par randomisation à un groupe de patients et le placebo à l’autre groupe, en plus des diurétiques et des IEC qui continuaient d’être prescrits dans les deux groupes. L’essai CIBIS II a montré que le bêta-bloquant, associé aux diurétiques et IEC, diminuait de 34 % le risque de décès dans l’insuffisance cardiaque par rapport au placebo associé aux diurétiques et IEC.
— L’essai doit être randomisé. Seule l’attribution aléatoire (donc par tirage au sort) des traitements dans chacun des groupes de patients qui font l’objet de la comparaison permet d’éviter toute sorte de biais de sélection qui risqueraient de fausser les résultats de l’essai. En l’absence de randomisation, c’est-à-dire de tirage au sort, l’attribution des traitements serait soumise aux effets d’une sélection consciente ou inconsciente effectuée par les médecins investigateurs.
Cette randomisation peut être stratifiée, c’est-à-dire qu’elle assure une répartition égale entre les groupes d’un facteur possiblement confondant pour les résultats de l’essai (par exemple le tabagisme dans les essais de prévention cardiovasculaire) — L’essai doit être réalisé, si possible, en double-aveugle. En effet si la randomisation des traitements entre les groupes de patients permet à l’inclusion de s’assurer de l’absence de différence significative initiale entre les groupes, il importe que pendant l’essai le principe du double-aveugle soit maintenu afin d’éviter toute subjectivité (du patient et de l’investigateur) dans la mesure des effets des traitements. On sait que lorsque le double-aveugle n’est pas possible, l’observation des effets d’un médicament par le médecin investigateur risque d’être biaisée et la propre conviction du patient peut également modifier les résultats. Il faut alors essayer de conduire l’essai au moins en simple aveugle afin de supprimer le biais subjectif lié au point de vue du patient. Si l’essai ne peut être mené qu’en ouvert (le patient et l’investigateur connaissent le traitement), la méthodologie PROBE [17] permet de faire évaluer en aveugle, par un observateur indépendant, les résultats de l’essai. Encore faut-il que les critères de mesure d’effet des traitements se prêtent à cette méthode, c’est à dire qu’ils soient ‘‘ externalisables ’’ pour permettre leur analyse par l’évaluateur indépendant ignorant la répartition individuelle des traitements.
— Les critères de mesure d’effet doivent être parfaitement définis. Dans tout essai comparatif, il convient de choisir un critère principal de mesure des effets des traitements. En effet, l’hypothèse principale qui est à l’origine de l’essai, débouche sur un objectif principal (ce que l’on veut confirmer) et donc sur la définition d’un seul critère de mesure d’effet qui doit être parfaitement relié à l’hypothèse et à l’objectif principaux. Ce critère doit être unique (par exemple la mortalité toutes causes confondues). Cependant il est de plus en plus fréquent d’observer, notamment dans les essais cliniques cardiovasculaires, l’utilisation de critères principaux dits ‘‘ composites ’’ qui associent plusieurs manifestations de la même maladie. Par exemple, il est courant de voir la mesure associée de plusieurs
complications de l’athérosclérose : mortalité cardiovasculaire + nombre d’infarctus du myocarde non létaux + accidents vasculaires cérébraux non létaux comme ceci a été réalisé dans l’étude CAPRIE [18] qui a montré la supériorité d’un anti-agrégant plaquettaire, le clopidogrel, sur l’aspirine chez des patients athéroscléreux. Certains y ajoutent même le nombre d’interventions non programmées de revascularisation cardiaque (pontage AC ou angioplastie) considérant qu’il s’agit d’évènements traduisant l’évolution d’une maladie arté- rielle unique, l’athérome. L’intérêt d’un critère composite est qu’il augmente le nombre d’évènements qui risquent de survenir pendant un temps donné, raccourcissant ainsi la durée de l’étude et augmentant la puissance statistique de l’essai (c’est-à-dire la probabilité de montrer une différence significative entre les traitements si elle existe). Encore faut-il qu’on associe bien dans le critère composite des évènements tels que la morbidité et la mortalité liées à une seule maladie, celle qu’on étudie, et non pas des évènements qui ne sont pas liés par une même causalité physiopathologique. Les autres critères de mesure d’effets d’un traitement sont dits ‘‘ secondaires ’’, c’est-à-dire qu’ils n’interviennent pas dans le calcul du nombre de patients à inclure dans l’essai. D’un point de vue médical ils peuvent être intéressants à mesurer mais, lorsqu’il n’existe pas de différence significative entre les deux traitements sur un critère secondaire de l’étude, cela ne signifie rien car la puissance statistique de l’essai n’a pas pris en compte les critères secondaires. Par exemple, dans un essai de prévention secondaire cardiovasculaire post-infarctus du myocarde, la différence entre deux traitements pourra être significative sur le critère principal (mortalité toutes causes confondues) mais seulement significative sur un des critères secondaires (accidents vasculaires cérébraux) et non sur un autre (récidive d’infarctus du myocarde). Ceci peut seulement suggérer qu’il aurait fallu inclure plus de malades dans l’essai pour montrer une différence significative sur le critère secondaire récidive d’infarctus du myocarde si cette différence existe. Ces critères secondaires, tout comme les comparaisons statistiques entre des sous-groupes de patients déterminés a posteriori, ont surtout l’intérêt d’attirer l’attention des investigateurs sur de nouvelles hypothèses à confirmer dans des essais cliniques ultérieurs.
— Le calcul des effectifs de patients à inclure dans l’essai contrôlé doit être explicité et vérifiable. Le protocole d’un essai comparatif (et l’article en rapportant les résultats) doit obligatoirement comporter un calcul des effectifs de patients à inclure dans l’étude. En effet le nombre des patients de l’essai n’est pas laissé au hasard. Il est calculé à partir d’éléments déterminés au préalable. Il faut en premier lieu savoir si l’essai comparatif est un essai de supériorité (l’objectif est alors de démontrer qu’un médicament est significativement supérieur au comparateur) ou s’il s’agit d’un essai d’équivalence (l’objectif est de démontrer que les deux traitements sont équivalents), qui se limite le plus souvent à l’objectif de démontrer que le nouveau médicament n’est pas inférieur au comparateur (après avoir défini une zone d’équivalence à l’intérieur de laquelle on admet l’équivalence des traitements). Ensuite le calcul du nombre des patients nécessite
d’avoir choisi le critère principal de mesure d’effet et de connaître sa variabilité ou sa variation spontanée qui apparaîtra dans le groupe contrôle pendant la durée de l’essai. Il faut enfin déterminer les risques statistiques de première et deuxième espèces qui sont acceptés dans l’essai. Le risque alpha est celui de dire qu’une différence entre deux traitements est significative alors qu’elle ne l’est pas.
Il est communément accepté à 5 % ce qui correspond à l’expression classique p< 0,05. Le risque bêta est celui de dire qu’une différence n’est pas significative alors qu’elle l’est en réalité. Le calcul du nombre de sujets permet de donner à l’essai clinique la puissance statistique suffisante pour montrer cette différence lorsqu’elle existe. Le risque bêta est communément accepté à 10 ou 20 % ce qui correspond à une puissance statistique de 0,90 ou 0,80 respectivement.
— Le plan expérimental de l’essai doit être adapté à l’objectif principal. La façon dont va se dérouler l’essai doit permettre d’atteindre l’objectif que l’on s’est fixé.
Très schématiquement, deux plans expérimentaux sont couramment utilisés.
L’un, appelé essai croisé, est un essai dans lequel tous les sujets de l’étude participent aux différentes phases de l’essai, et c’est l’ordre des phases qui est tiré au sort pour chaque sujet. On dit que dans ce type d’essai le patient est ‘‘ son propre témoin ’’ car il prend le médicament testé et le comparateur. L’autre, appelé essai en groupes parallèles, est un essai dans lequel les sujets sont attribués par tirage au sort à l’un des groupes de l’étude et y resteront jusqu’à la fin de l’essai. Ces deux types d’essais ne répondent pas aux mêmes objectifs et ont chacun leurs avantages et leurs inconvénients. L’essai croisé par exemple n’est utilisable que si l’état pathologique des patients est stable du début à la fin de l’essai et si notamment s’il est identique au début de chacune des phases de traitement, ce qui nécessite des périodes transitoires de retour à l’état initial pour les patients entre chaque phase. Par ailleurs il faudra vérifier que les effets d’un traitement sont de même intensité que ce traitement soit administré en première ou en deuxième période de l’essai. L’avantage de l’essai croisé, est qu’il nécessite d’inclure un faible nombre de patients puisque ce sont ces mêmes patients qui participent à toutes les phases de l’essai, d’où une faible variabilité intraindividuelle. L’essai en groupes parallèles ne souffre pas de ces problèmes mais il nécessite d’inclure un plus grand nombre de patients car la variabilité interindividuelle y est évidemment plus importante que dans l’essai croisé où les patients sont leur propre témoin.
— Enfin, la population des patients sur laquelle portera l’analyse des résultats de l’essai doit être précisée. L’analyse statistique la plus robuste est dite ‘‘ en intention de traiter ’’ c’est-à-dire qu’elle concerne tous les patients inclus dans l’essai, dès leur randomisation. Certains essais utilisent une analyse en intention de traiter modifiée, c’est-à-dire qu’elle ne concerne que les patients randomisés qui ont pris au moins une dose du médicament ou qui ont été réexaminés au moins une fois après leur randomisation. L’autre type d’analyse statistique est appelée analyse per-protocole, c’est-à-dire qu’elle ne tient compte que des patients qui ont été conformes au protocole de l’étude depuis leur inclusion
jusqu’à la fin. Cette analyse aboutit donc à une sorte de sélection des patients analysés, ce qui peut introduire des biais dans la significativité des résultats. En fait, lorsque l’essai contrôlé a été correctement réalisé, et lorsqu’il y a très peu ou pas de patients perdus de vue avant la fin de l’essai, les résultats des deux analyses, en intention de traiter et per-protocole, ne différent pas. S’ils différent, c’est l’analyse en intention de traiter qui sera retenue car elle est finalement plus conservatrice et plus robuste que l’analyse per-protocole.
Les essais cliniques contrôlés doivent donc répondre à ces exigences méthodologiques principales pour être pris en compte dans les recommandations thérapeutiques. Le niveau de preuve qu’ils apportent est aujourd’hui le plus élevé et le moins discutable.
INTÉRÊT ET LIMITES DES ESSAIS CLINIQUES CONTRO
Ù
LÉS
Les résultats des essais cliniques contrôlés font l’objet aujourd’hui de nombreuses communications auprès des médecins et sont pris en compte dans les recommandations thérapeutiques écrites par les autorités de santé, comme la Haute Autorité de Santé (HAS) ou l’Agence Française de Sécurité Sanitaire des Produits de Santé (AFSSAPS), les académies, les sociétés savantes, ou les conférences de consensus.
L’intérêt de ces résultats pour la pratique médicale est aussi important que les résultats d’un essai soient positifs (confirmation de l’hypothèse de départ) ou négatifs (non confirmation de l’hypothèse). Ceci est particulièrement vrai dans le domaine des essais de médicaments, bien que les firmes pharmaceutiques privilé- gient plus souvent la communication sur les résultats positifs que sur les résultats négatifs de leurs médicaments. Citons par exemple les résultats négatifs de l’étude Field [19] menée chez 9 795 patients diabétiques de type 2, selon une méthodologie correcte, et qui montrent qu’un fibrate, le fenofibrate, prescrit à des diabétiques hypercholestérolémiques, ne diminue pas significativement le risque de survenue d’un infarctus du myocarde létal ou non. Citons, très récemment les résultats de l’étude RUTH [20] qui montrent qu’un modulateur des récepteurs aux œstrogènes, le raloxifène utilisé dans le traitement de l’ostéoporose post-ménopausique, ne prévient pas chez les femmes ménopausées le risque d’accident coronarien et n’a pas d’effet significatif sur la mortalité globale, bien qu’il diminue significativement le risque des cancers du sein oestrogéno-dépendants. Ces résultats négatifs, tout comme les résultats positifs, doivent aboutir à des modifications de la pratique médicale.
Cependant, même si les preuves d’efficacité apportées par les essais cliniques contrôlés sont indiscutables, il ne faut pas méconnaître les limites de ces situations expérimentales qui sont parfois éloignées de la pratique médicale usuelle. C’est toute la difficulté de transposer dans la médecine de tous les jours les résultats des essais cliniques contrôlés. Plusieurs raisons peuvent expliquer ces difficultés (tableau no 3).
TABLEAU no 3. — Limites des résultats des essais cliniques contrôlés — Effectifs limités de patients exposés au médicament — Sélection rigoureuse des patients et exclusion des formes pathologiques atypiques ou des polypathologies — Exclusion des associations médicamenteuses risquant de modifier les résultats des essais — Conditions particulières de surveillance clinique et biologique des patients au cours des essais — Durée limitée des traitements dans les essais La première limite des essais cliniques contrôlés vient du faible nombre de patients inclus dans les essais par rapport à la population totale des patients qui seront ensuite exposés au traitement. Par exemple, même si 2 000 à 5 000 personnes ont participé aux différentes phases des essais cliniques d’un nouveau médicament avant son Autorisation de Mise sur le Marché (AMM), cela peut ne représenter qu’une infime proportion du nombre des patients qui seront traités par le médicament lorsqu’il aura été commercialisé. Du coup, la valeur prédictive des essais est limitée notamment dans le domaine des effets indésirables des médicaments. Si la probabilité de survenue d’un effet indésirable grave est faible, il faudra un grand nombre de patients exposés au médicament pour avoir une chance d’observer cet effet. Ceci justifie les études pharmaco-épidémiologiques post-AMM de cohortes qui permettent d’observer un grand nombre de patients exposés au traitement et peuvent permettre de détecter un effet indésirable rare. La seconde limite des essais cliniques découle de la sélection rigoureuse des patients lors de leur inclusion dans les essais.
Cette sélection est justifiée par la nécessité d’éliminer des facteurs confondants tels que les formes atypiques de la maladie ou les poly-pathologies, qui risqueraient de masquer la différence entre les traitements que l’on veut montrer dans l’essai.
L’autre inconvénient d’une grande variabilité interindividuelle des patients de l’essai est d’augmenter le nombre des patients nécessaires pour mettre en évidence une différence significative entre les traitements, si elle existe. En conséquence, les patients inclus dans les essais, soigneusement sélectionnés à l’entrée, risquent de ne pas être représentatifs de la majorité des patients qui seront traités par le médicament par la suite.
Dans le même ordre d’idée, il est habituel d’interdire dans les essais cliniques l’association aux médicaments de l’essai d’autres médicaments qui risquent pourtant ensuite d’être prescrits aux patients en même temps que le nouveau médicament (lorsque ce n’est pas le patient lui-même qui décide d’associer le nouveau médicament à un médicament détenu dans la pharmacie familiale). Même si des essais cliniques spécifiques sont toujours demandés pour étudier les effets des associations médicamenteuses les plus probables, il est possible que, dans la ‘‘ vraie vie ’’, des
associations médicamenteuses non étudiées et non prévisibles, entraînent des risques pour la santé du patient.
La quatrième limite des essais cliniques contrôlés concerne les conditions même de conduite des essais. Dans les essais, les médecins investigateurs doivent suivre à la lettre le déroulement de l’essai prévu par le protocole. La surveillance clinique et biologique des patients y est soigneusement organisée et il est courant de considérer que le patient lui-même, ayant accepté de participer à l’essai, respectera plus scrupuleusement les conseils du médecin. Or ce n’est pas toujours le cas dans la ‘‘ vraie vie ’’ où le suivi d’un patient peut être irrégulier et où n’existe pas la contrainte d’un protocole à respecter.
Enfin, la durée des traitements dans les essais cliniques est forcément limitée dans le temps. Or lorsqu’il s’agit d’une pathologie chronique dans laquelle le médicament risque d’être prescrit pendant plusieurs années, la durée limitée des essais ne permet pas de prédire à coup sûr le maintien d’une efficacité et d’une tolérance semblables dans le long terme à ce qu’elles étaient dans le court terme des essais cliniques.
Ces cinq limitations principales des essais cliniques contrôlés doivent être connues et prises en compte. Si elles n’enlèvent rien au niveau de preuve apporté par les essais, elles justifient que d’autres types d’études soient poursuivis lorsque le médicament est mis sur le marché et est largement prescrit. Il s’agit alors des études observationnelles pharmaco-épidémiologiques consistant le plus souvent à suivre pendant une longue période une cohorte de patients exposés au nouveau médicament. Ces études sont maintenant demandées par l’HAS lorsqu’elle estime que les essais cliniques pré-AMM, bien que permettant la mise sur le marché du médicament, ne sont pas totalement suffisants pour prédire dans la pratique médicale usuelle l’acceptabilité du traitement et sa tolérance au long cours. Ces études peuvent également servir à mieux cerner les caractéristiques des patients exposés au médicament.
Enfin, il convient de s’intéresser à un problème souvent ignoré. Certains domaines de la recherche médicale sont totalement abandonnés car ils n’intéressent pas l’industrie ou parce qu’ils posent des problèmes de santé publique qui n’ont pas à être résolus par des investisseurs privés. Or certaines de ces questions thérapeutiques sont importantes et restent aujourd’hui sans réponse. Par exemple, il a fallu attendre les années 2000 pour que le Medical Research Council anglais prenne l’initiative de tester sur une grande échelle (plus de 10 000 patients) les effets d’une pratique médicale courante depuis des dizaines d’années, la prescription de corticoïdes intraveineux après un traumatisme crânien. L’essai CRASH [21] contrôlé randomisé versus placebo a montré que la mortalité ou le degré des séquelles neurologiques chez ces patients n’étaient pas significativement améliorés par le traitement et qu’il y avait même une augmentation de près de 20 % de la mortalité dans les deux premières semaines de traitement. De même, c’est à l’initiative du National Heart, Lung and Blood Institute américain qu’a été réalisée l’étude AFFIRM comparant chez plus de 4 000 patients en fibrillation auriculaire deux stratégies thérapeutiques, médicaments anti-arythmiques versus médicaments ralentisseurs de la fréquence
ventriculaire (digoxine, bêta-bloquants ou antagonistes calciques) [22]. Après un suivi moyen de trois ans et demi, l’essai montre qu’il n’existe pas de différence significative entre les deux stratégies thérapeutiques sur la mortalité toutes causes des patients. Dans un autre domaine, l’hypertension artérielle, problème majeur de santé publique, il a fallu attendre l’étude ALLHAT publiée en 2000 [23] et 2002 [24] pour montrer que, chez des hypertendus ayant un facteur de risque coronarien, le traitement de l’hypertension artérielle devait débuter par un diurétique mais que les autres traitements proposés, comme un antagoniste calcique ou un inhibiteur de l’enzyme de conversion de l’angiotensine, partageaient avec le diurétique les mêmes effets de prévention des accidents coronariens fatals ou non fatals et réduisaient la mortalité. En revanche un médicament alpha-bloquant augmentait significativement le risque d’insuffisance cardiaque.
Ces différents exemples d’essais cliniques contrôlés montrent qu’aujourd’hui, les praticiens auront de plus en plus besoin de ces essais à promotion institutionnelle pour résoudre des questions de choix thérapeutique. De plus en plus d’essais cliniques contrôlés devront ainsi être mis en place à l’instigation et/ou avec le financement d’instances publiques à l’image de ce que font le NIH, le National Heart Lung and Blood Institute ou le National Cancer Institute aux USA et le Medical Research Council anglais. Ces essais cliniques contrôlés permettront de comparer des stratégies thérapeutiques ou des traitements qui n’ont pas encore été comparés et devant le choix desquels le médecin prescripteur reste perplexe. Le fait de réaliser ces essais en France ou au moins en Europe aurait également l’avantage d’inclure des populations correspondant parfaitement aux patients français, ce qui n’est pas forcément le cas des populations des Etats-Unis qui n’ont pas exactement les mêmes facteurs de risque, notamment cardiovasculaire, que les patients français.
Malheureusement aucune institution publique française ne peut financer à elle seule aujourd’hui de tels essais cliniques multicentriques de grande ampleur dont le coût correspond à plusieurs centaines de milliers d’euros.
CONCLUSION
Il semble donc clairement établi aujourd’hui que l’essai clinique contrôlé randomisé constitue le moyen le plus sûr et le plus efficace pour apporter aux médecins des résultats crédibles et fiables qui leur permettent de rationaliser leurs pratiques diagnostiques et thérapeutiques. La médecine factuelle qui repose sur l’analyse et la prise en compte des résultats de ces essais constitue aujourd’hui le fondement de l’enseignement de la pratique médicale dans les facultés de médecine. Encore faut-il que des promoteurs privés et/ou institutionnels mettent en place de tels essais et que leurs résultats soient largement diffusés et intégrés à des recommandations thérapeutiques officielles que devront suivre les médecins prescripteurs.
BIBLIOGRAPHIE [1] LIND J. —
A treatise of the scurvy in three parts. Containing an inquiry into the nature causes and cure of that disease, together with a critical and chronological view of what has been published on the subject. A. Millar, London, 1753.
[2] MARTINI E. — Comment Lind n’a pas découvert le traitement contre le scorbut. Histoires des
Sciences Médicales , 2005, tome 39, no 1 , 79-92.
[3] LOUIS P.C.A. —
Recherches sur les effets de la saignée . 1835, Paris, De Mignaret Editeur.
[4] Medical Research Council — Streptomycin treatment of pulmonary tuberculosis.
Brit. Med. J ., 1948, ii , 769-82.
[5] BOISSEL J.P. — Impact of randomized clinical trials on medical practices.
Control. Clin. Trials , 1989, 10 , 120 S-134 S.
[6] BOUVENOT G. et VRAY M. — E ssais cliniques. Théorie, pratique et critique. p. XV 4ème édition.
Médecine Sciences Flammarion, Paris, 2006.
[7] Évidence — Based Medicine Working Group — Evidence — based medicine. A new approach to teaching the practice of medicine. J.A.M.A ., 1992, 268 , 2420-5.
[8] COOK D.J., GUYATT G.H., LAUPACIS A., SACKETT D., GOLDBERG R.J. — Clinical recommendations using levels of evidence for antithrombotic agents. Chest , 1995, 108 , 227 S-230 S.
[9] RUBERMAN W., WEINBLATT E., GOLDBERG J.D., FRANK C.W., SHAPIRO S. — Ventricular premature beats and mortality after myocardial infarction. N. Engl. J. Med. , 1977, 297 , 750-7.
[10] MOSS A.J., DAVIS H.T., DECAMILLA J., BAYER L.W. — Ventricular ectopic beats and their relation to sudden and nonsudden cardiac death after myocardial infarction. Circulation , 1978, 60 , 998-1003.
[11] BIGGER J.T JR, FLEISS J.L., KLEIGER K., MILLER J.P., ROLNITZKY L.M. and The Multicenter Post Infarction Research Group — The relationship between ventricular arrythmias, left ventricular dysfunction and mortality in the two years after myocardial infarction. Circulation , 1984, 69 , 250-8.
[12] MUKHARJI J., RUDE R.E., POOLE W.K., GUSTAFSON N., THOMAS L.J. JR, STRAUSS H.W. et al —
The Milis Study Group Risk factors for sudden death after acute myocardial infarction :
two-year follow-up. Am. J. Cardiol ., 1984, 54 , 31-6.
[13] The Cardiac Arrhythmia Pilot Study (CAPS) investigators — Effects of encainide, flecainide, imipramine and moricizine on ventricular arrhythmias during the year after acute myocardial infarction : The CAPS. Am. J. Cardiol ., 1988, 61 , 501-9.
[14] The Cardiac Arrhythmia Suppression Trial (CAST) Investigators — Preliminary report : Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction . N. Engl. J. Med ., 1989, 321 , 406-12.
[15] ECHT D., LIEBSON P.R., BRENT MICHELL L., PETERS R.W., OBIAS-MANNO D., BARKER A.H. et al — Mortality and morbidity in patients receiving encainide, flecainide or placebo. The Cardiac
Arrhythmia Suppression Trial.
N. Engl. J. Med., 1991, 324 , 781-8.
[16] CIBIS II investigators and committees — The Cardiac insufficiency Bisoprolol Study II (CIBIS II). Lancet , 1999, 353 , 9-13.
[17] HANSSON L., HEDNER T., DAHLOF B. — Prospective Randomized Open Blinded-Endpoint (PROBE) Study : a novel design for intervention trials. Blood Pressure , 1992, 1 , 113-9.
[18] CAPRIE Steering committee — A randomized,blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet , 1996, 348 , 1329-39.
[19] The FIELD Study Investigators — Effect of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD Study) : randomised controlled trial. Lancet , 2005, 366 , 1849-61.
[20] BARRETT-CONNOR E., MOSCA L., COLLINS P., GEIGER M.J., GRADY D., KORNITZER M. et al . for the Raloxifene Use for the Heart (RUTH) Trial investigators. — Effects of Raloxifene on cardiovascular events and breast cancer in postmenopausal women. N. Engl. J. Med., 2006, 355 , 125-37.
[21] CRASH Trial collaborators — Effect of intravenous corticosteroids on death within 14 days in 10 008 adults with clinically significant head injury (MRC CRASH trial) : randomised placebocontrolled trial. Lancet , 2004, 364 , 1321-28.
[22] The Atrial Fibrillation Follow-up investigation of Rhythm Management (AFFIRM) investigators — A comparison of rate control and rhythm control in patients with atrial fibrillation. N.
Engl. J. Med ., 2002, 347 , 1825-33.
[23] The Antihypertensive and lipid-lowering treatment to prevent Heart Attack Trial (ALLHAT) — Major cardiovascular events in hypertensive patients randomized to doxasocin vs chlorthalidone. J.A.M.A., 2000, 283 , 1967-75.
[24] The Antihypertensive and lipid-lowering treatment to prevent Heart Attack Trial (ALLHAT) — Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic. J.A.M.A ., 2002, 288 , 2981-97.
DISCUSSION
M. Jean-Paul GIROUD
Après les essais cliniques nécessaires pour obtenir l’AMM d’un nouveau médicament, est-il nécessaire, à votre avis, de poursuivre les essais cliniques ?
Les essais cliniques sont indispensables pour pouvoir répondre aux questions de la commission d’AMM concernant la démonstration du rapport bénéfice-risque du nouveau médicament. Cependant de plus en plus souvent, la Haute Autorité de Santé et la Commission de la Transparence demandent aux industriels de mettre en place après l’AMM des études pharmaco-épidémiologiques qui ne sont pas des essais cliniques randomisés contrôlés comme ceux que je viens de décrire. Ces études observationnelles de cohortes de patients ont plusieurs objectifs : décrire la façon dont le médicament est prescrit par les médecins, observer les caractéristiques des patients qui vont être exposés au médicament, détecter la survenue d’effets indésirables rares qui auraient pu ne pas être détectés avant l’AMM et analyser les risques liés à des associations médicamenteuses potentiellement dangereuses. L’industriel doit d’ailleurs avoir déposé à l’AFSSAPS en même temps que son dossier d’AMM, un plan de gestion de risque, afin de prévenir le mieux possible les risques d’événements indésirables lorsque le médicament est mis sur le marché.
M. Roger NORDMANN
Malgré leurs limites, vous avez parfaitement démontré l’intérêt des essais cliniques contrô- lés. Il nous semble cependant que le grand public ignore la différence entre de tels essais
(correspondant à la médecine factuelle, terme beaucoup moins bien compris que ‘‘ évidence based medicine ’’) et les publications portant sur un nombre restreint (et non défini scientifiquement) de patients. L’Académie ne devrait-elle pas recommander aux journalistes (notamment médicaux) de bien différencier les résultats des essais cliniques contrôlés (à diffuser largement) et les autres résultats (dont la diffusion est souvent injustifiée ou prématurée) ?
Je ne peux qu’être d’accord avec cette proposition car il est important que les journalistes puissent véritablement faire la différence entre diverses études cliniques en fonction de leur niveau de preuve respectif. Il y a ainsi une différence fondamentale entre les informations apportées par un essai clinique contrôlé randomisé (qui apporte aujourd’hui le meilleur niveau de preuve) et celles apportées par une étude ouverte non randomisée et sans groupe témoin (dont le niveau de preuve est très faible). Ceci devrait être clairement pris en compte par les journalistes afin que les informations qu’ils transmettent aient la qualité et l’objectivité qu’on peut attendre aujourd’hui.
M. Pierre GODEAU
Un essai négatif coordonne-t-il à priori toute tentation thérapeutique analogue ? Si on se réfère à l’essai CAST que vous avez cité, quel a été le mécanisme de l’échec ? Les extrasystoles fréquentes dont l’incidence pronostique fâcheuse a été bien établie n’étaientelles qu’un indicateur d’un état myocardique de terrain et non un facteur causal de mortalité ? Une sélection plus précise du type d’ES aurait-elle pu modifier les résultats ? L’échec des anti-arythmiques est-il dû à une autre action, qu’à leur seul rôle anti-arythmique ?
Si l’on reprend les résultats de l’étude CAST, on peut considérer que l’importance qu’ils ont eu à l’époque vient du fait que les cardiologues avaient « l’intime conviction » que supprimer les extrasystoles ventriculaires entraînait automatiquement une amélioration du pronostic des patients puisqu’on avait montré que leur présence aggravait ce pronostic. En fait, il fallait introduire dans ce raisonnement le risque potentiel des antiarythmiques. On sait maintenant qu’en présence d’une ischémie myocardique, des antiarythmiques de classe I (bloqueurs des canaux sodiques des cellules myocardiques). comme ceux utilisés dans l’étude CAST peuvent aggraver la situation et déclencher des arythmies létales (tachycardies et fibrillations ventriculaires). C’est pourquoi ils ne sont plus indiqués dans cette pathologie alors qu’ils peuvent être bien tolérés dans le traitement d’arythmies sur myocarde non ischémique (comme la fibrillation auriculaire). Les résultats de l’étude CAST proviennent d’une évaluation insuffisante du risque de ces médicaments en situation d’ischémie myocardique.
M. Roger BOULU
Qu’en est-il de la classe des médicaments homéopathiques ?
Il n’existe que très peu de réels essais cliniques randomisés contrôlés effectués pour tester des médicaments homéopathiques. Les études homéopathiques sont généralement ouvertes, sans groupe témoin et sans randomisation et donc ne permettent pas de conclure quant à l’efficacité de ces médicaments. La situation est encore plus difficile si la prescription homéopathique est individualisée. Dans ce cas, on ne peut pas utiliser la randomisation et les biais subjectifs sont énormes.
M. Pierre BÉGUÉ
Les deux populations des malades recrutés pour les essais sont étudiées en intention de traiter (ITT) et en préprotocole (PPE), pour celles qui ont complètement suivi le protocole.
Les vaccins posent-ils un problème plus spécifiques, en particulier par l’intérêt de la population PPE ? En effet, la recommandation d’un vaccin doit être très précise sur les doses et sur leur espacement.
L’avantage de l’analyse des résultats en ITT est qu’ils tiennent compte des déviations ou des violations de protocole. On considère donc que si, malgré ces imperfections ou ces erreurs, le résultat de l’essai est statistiquement significatif, il est crédible et authentique.
Cette analyse est donc considérée comme plus « robuste » et plus fiable que celle PPE où on n’analyse que les patients conformes au protocole ce qui aboutit en fait à une sélection d’un groupe de patients particuliers. Concernant les vaccins, le même raisonnement s’applique aux essais de médicaments.
M. Raymond ARDAILLOU
Quand doit-on interrompre un essai randomisé ?
Il s’agit d’un problème éthique. Puisque tout essai, comme toute situation expérimentale, comporte des risques pour les patients, on doit interrompre un essai chaque fois que, soit le résultat attendu est obtenu, soit le résultat n’a aucune chance d’être obtenu, soit l’existence d’effets indésirables graves dans un des groupes de patients impose d’arrêter l’essai pour garantir la sécurité des patients.
* Pharmacologie, Hôpital Saint-Antoine AP-HP et Faculté de Médecine Pierre et Marie Curie. Site Saint-Antoine, Université Paris 6, 27 rue de Chaligny 75012 Paris. mail : patrice.jaillon@upmc.fr Tirés-à-part : Professeur Patrice JAILLON, même adresse Article reçu le 6 février 2007et accepté le 12 mars 2007.
Bull. Acad. Natle Méd., 2007, 191, nos 4-5, 739-758, séance du 3 avril 2007