
Résumé
Depuis 2005, des affections neurologiques (porencéphalie, leucoencéphalopathie, hémorragies intracérébrales sans hypertension) et des tortuosités des artérioles rétiniennes ont été rapportées en association avec des mutations du gène COL4A1 qui code pour la chaîne alpha 1 du collagène de type IV, principal constituant des membranes basales. L’étude de trois familles nous a permis de décrire un nouveau syndrome que nous avons appelé AHNAC pour angiopathie héréditaire avec néphropathie, anévrismes et crampes musculaires et qui est associé à des altérations morphologiques des membranes basales dans le rein et dans la peau. Cette nouvelle « basalopathie » est la conséquence de mutations qui remplacent une glycine par un autre acide aminé dans les exons 24 et 25 de COL4A1. Les corrélations phénotype-génotype sont discutées ainsi que la place du syndrome AHNAC dans le diagnostic des formes autosomiques dominantes d’hématurie, de multikystose rénale, d’anévrismes intracrâniens, et de crampes musculaires. * Membre correspondant de l’Académie nationale de médecine ** Néphrologie et Dialyses. Unité INSERM UMR S 702. UPMC. Hôpital Tenon, Paris Tirés à part : Professeur Pierre Ronco, même adresse Article reçu le 17 décembre 2007, accepté le 11 février 2008
Summary
Since 2005, several neurological diseases, including porencephaly, leukoencephalopathy and intracerebral hemorrhage without hypertension, and retinal arteriolar tortuosity have been linked to mutations in the COL4A1 gene, which encodes the alpha1 chain of type IV collagen, the main constituent of basement membranes. In three families, we observed a new syndrome that we called HANAC, for hereditary angiopathy with nephropathy, aneurysms and muscle cramps, which is associated with morphological alterations of cutaneous and renal basement membranes. This novel ‘‘ basalopathy ’’ is caused by glycine mutations in
COL4A1 exons 24 and 25. We discuss phenotype-genotype correlations and the implications of the HANAC syndrome for the diagnosis of autosomal dominant hematuria, cystic kidney disease, intracranial aneurysms, and muscle cramps.
INTRODUCTION
Nous avons récemment rapporté dans le
New England Journal of Medicine, trois familles atteintes d’un nouveau syndrome à transmission autosomique dominante que nous avons appelé AHNAC pour a ngiopathie h éréditaire avec n éphropathie, a névrismes et c rampes musculaires [1]. Ce syndrome est associé à des mutations hétérozygotes du gène
COL4A1 qui code pour la chaîne α1 du collagène de type IV, principal constituant des membranes basales (MB) de l’organisme. Les altérations morphologiques des MB, que nous avons identifiées dans le rein et dans la peau, sont très probablement à l’origine des manifestations cliniques observées dans les autres organes. Elles contribuent à la définition de cette nouvelle « basalopathie ».
Les objectifs de cette lecture sont de replacer le syndrome AHNAC dans le contexte des maladies héréditaires des MB associées à des mutations des gènes des différentes chaînes du collagène IV, de préciser les manifestations du syndrome, de discuter les corrélations génotype-phénotype, et de définir les conditions dans lesquelles des mutations du gène COL4A1 devraient être recherchées.
MEMBRANES BASALES ET COLLAGENE IV
Les MB constituent une matrice extracellulaire spécialisée, qui s’interpose entre les cellules et le tissu conjonctif sous-jacent. Elles renferment trois types de macromolécules : les glycoprotéines collagéniques essentiellement représentées par le collagène de type IV, mais aussi les collagènes VI, XV et XVIII ; les glycoprotéines non collagéniques parmi lesquelles les laminines, l’entactine/nidogène, la fibronectine, les fibulines, SPARC (« Secreted Protein Acidic, Cystein Rich ») ; les protéoglycans à chaînes sulfatées, essentiellement le perlécan et l’agrine [2]. Les molécules de collagène IV d’une part, et les trimères de laminines d’autre part, forment dans l’espace extracellulaire deux réseaux moléculaires distincts, réunis grâce à leur interaction avec l’entactine/nidogène. Par ailleurs, certains constituants de la MB interagissent avec des molécules d’adhérence exprimées à la surface des cellules adjacentes, en particulier certaines intégrines, les récepteurs DDR (« Discoidin Domain Receptors ») et le complexe dystroglycan [2]. L’expression restreinte à certains tissus d’isoformes de collagène IV, de laminines, et de protéoglycans rend compte de la diversité moléculaire et fonctionnelle des MB selon les organes et selon le stade de développement [3].
Le collagène de type IV est le collagène majeur des MB. Il se décline en six isoformes, représentées par les chaînes α1(IV), α2(IV), α3 (IV), α4(IV), α5(IV) et α6(IV). Ces chaînes partagent une importante homologie de séquence et d’organisation structurale [4]. Elles sont formées de trois domaines : un large domaine collagénique d’environ 1 400 résidus, constitué d’une répétition du triplet Gly-X-Y, X et Y correspondant à divers acides aminés (aa), interrompue par une vingtaine de courtes séquences non collagéniques ; un domaine C-terminal non collagénique de structure globulaire d’environ 230 aa, dit domaine NC-1 ; un domaine N-terminal de 15 à 25 aa, dit domaine 7S. Chaque molécule de collagène IV est un hétérotrimère (ou protomère) formé de trois chaînes α. L’interaction spécifique entre les domaines NC1 des chaînes α initie la trimérisation et précède la formation d’une triple hélice entre les domaines collagéniques, au sein de laquelle les résidus glycine jouent un rôle stabilisateur essentiel. Les six chaînes de collagène IV ne produisent que trois types d’hétérotrimères formant des réseaux, respectivement α1α1α2(IV), α3α4α5(IV) et α5α5α6(IV) [5]. L’expression du trimère α1α1α2(IV) est quasi ubiquitaire dans l’organisme, alors que les trimères α3α4α5(IV) et α5α5α6(IV) ont une expression tissulaire restreinte.
Ainsi, celle du trimère α3α4α5(IV) est limitée à certains organes : rein, cochlée, œil, poumon, testicule. Dans le rein, le trimère α3α4α5(IV) remplace le trimère α1α1α2(IV) pendant l’embryogenèse de la membrane basale glomérulaire (MBG) [3], tandis que les MB des tubules et de la capsule de Bowman sont composées principalement d’un mélange de trimères α1α1α2 (IV) et α5α5α6 (IV). Une fois sécrétées dans la matrice extracellulaire, les molécules de collagène IV s’organisent pour former des réseaux grâce à des interactions intermoléculaires multiples (Figure 1).
Fig. 1. — Représentation schématique des interactions entre molécules de collagène IV dans les MB.
Les gènes codant pour les six chaînes de collagène IV sont distribués en paire sur trois chromosomes différents, où ils sont disposés « tête à tête ». Chez l’homme, les gènes COL4A1 et COL4A2 sont présents sur le chromosome 13, les gènes COL4A3 et
COL4A4 sur le chromosome 2, et les gènes COL4A5 et COL4A6 sur le chromosome X.
SYNDROME D’ALPORT : PREMIÈRE MALADIE GÉNÉTIQUE DU COLLAGÈNE IV
La présence d’anomalies ultrastructurales de la MBG chez les patients ayant un syndrome d’Alport (SA) suggérait que le défaut moléculaire responsable de cette maladie affectait l’un de ses constituants essentiels. Cette hypothèse a été confirmée au cours des années 1990 par l’identification de mutations du gène COL4A5 dans la forme de SA liée à l’X [6], et des gènes
COL4A3 , COL4A4 au cours du SA autosomique récessif [7]. Plus récemment, des mutations hétérozygotes de
COL4A3 et COL4A4 ont été rapportées au cours de formes dominantes de SA et dans environ 50 % des familles présentant une hématurie familiale bénigne (HFB) [8].
Les progrès de l’analyse biochimique des MB, et l’étude des caractéristiques cliniques, génétiques et morphologiques au cours du SA, ont apporté des informations essentielles sur la biologie du collagène IV. Ainsi, le phénotype strictement glomé- rulaire, auditif et ophtalmologique observé au cours du SA est en accord avec l’expression restreinte du réseau α3α4α5 de collagène IV dans les différentes MB de l’organisme [9]. Par ailleurs, l’absence complète d’expression des chaînes α3(IV), α4(IV) et α5(IV) dans la MBG au cours des SA liés à l’X ou récessifs illustre l’interdépendance de chacune de trois chaînes de collagène IV pour la formation et l’intégration d’un réseau α3α4α5 fonctionnel dans la MBG, le défaut de l’une des chaînes n’étant pas substituable. L’absence complète de formation de l’hétérotrimère α3α4α5 explique la persistance définitive du réseau α1α1α2 dans la MBG adulte. La sensibilité accrue aux protéases de ce réseau collagénique, et sa moindre résistance aux contraintes liées au processus de filtration glomérulaire, pourraient en partie expliquer les anomalies de la MBG au cours du SA [10, 11].
Les anomalies des gènes COL4A3 , COL4A4 , et COL4A5 identifiées au cours du SA se répartissent sur l’ensemble de chacun des gènes et sont de nature diverse. Les mutations faux sens pathogènes affectent quasi exclusivement les résidus glycine du domaine collagénique, altérant ainsi de manière significative la stabilité conformationnelle de la triple hélice. Des corrélations phénotype/génotype n’ont été établies qu’au cours du SA lié à l’X. Ainsi, la présence de larges réarrangements géniques de COL4A5 apparaît associée à une progression plus rapide de la maladie rénale vers le stade terminal chez les hommes [12, 13], et à un risque accru de développer une insuffisance rénale terminale (IRT) chez les femmes hémizygotes [14]. Les mutations des gènes COL4A3 et COL4A4 chez les sujets hétérozygotes peuvent être totalement asymptomatiques, ou se manifester par une hématurie familiale bénigne (HFB) qui associe typiquement une hématurie isolée non évolutive et un amincissement régulier des MBG en microscopie électronique, ou encore induire la forme dominante de SA, où l’hématurie est associée à une protéinurie, voire à une insuffisance rénale évolutive, et à des anomalies auditives. Les effets phénotypiques différents de mutations voisines ou similaires restent mal compris, faisant évoquer le rôle de gènes modificateurs, de polymorphismes associés ou de facteurs environnementaux.
ANOMALIES DU GÈNE COL4A1 ET MALADIES DES PETITES ARTÈRES
CÉRÉBRALES
Contrairement aux réseaux α3α4α5(IV) et α5α5α6(IV), le réseau α1α1α2 est ubiquitaire et apparaît précocement au cours du développement embryonnaire [3]. L’invalidation combinée des gènes Col4a1 et Col4a2 chez la souris est létale chez l’embryon homozygote à dix jours et demi de développement [15]. Toutefois, l’ébauche d’une MB constituée d’un réseau de laminine et de nidogène est observée chez ces animaux, et les stades initiaux de l’organogénèse et de la vasculogenèse apparaissent préservés jusqu’à neuf jours de développement. L’absence d’intégration des molé- cules de collagène IV semble délétère à un stade où les MB sont soumises à des contraintes mécaniques plus importantes, en particulier dans le placenta et les vaisseaux [15]. Le phénotype des animaux hétérozygotes est néanmoins normal, indiquant l’absence d’effet néfaste de l’haploinsuffisance.
C’est en 2005 que les premières mutations du gène COL4A1 ont été rapportées.
Gould et coll caractérisent une souche de souris qui développent une maladie neurologique caractérisée par la survenue d’hémorragies cérébrales massives durant la période néonatale, à l’origine de larges cavités encéphaliques séquellaires et d’une forte mortalité néonatale [16]. Une mutation ponctuelle affectant une zone d’épissage et responsable d’une délétion complète de l’exon 40 du gène Col4a1 est identifiée dans cette souche murine [16]. Le phénotype présenté par ces animaux rappelle une pathologie héréditaire humaine à transmission autosomique dominante, la porencéphalie. Cette maladie est caractérisée par l’existence de larges cavités intracérébrales séquellaires d’hémorragies intracérébrales néonatales. Les manifestations cliniques sont souvent très sévères, associant déficits moteurs, retard mental, comitialité, avec parfois décès précoce des enfants. Dans deux familles présentant une porencéphalie et étudiées par Gould et coll [16], une liaison de la maladie est établie avec le locus COL4A1/COL4A2 en 13q34. Le séquençage du gène
COL4A1 confirme dans chacune des familles, la présence d’une mutation siégeant respectivement dans les exons 28 et 43 du gène, les deux mutations étant responsables de la substitution d’un résidu glycine dans le domaine collagénique [16]. Trois nouvelles mutations sont identifiées la même année, dont deux, localisées dans les exons 39 et 48, substituent une glycine, alors que la troisième affectant le codon initiateur ATG, a un impact inconnu sur la synthèse de la protéine [17]. Bien que ces cinq familles aient été repérées par la présence d’une porencéphalie affectant au moins l’un de leurs membres, l’analyse phénotypique révèle une grande diversité clinique de l’atteinte neurologique, aussi bien dans sa sévérité que dans son mode lésionnel. En effet, chez certains patients, l’apparition de la symptomatologie clini- que neurologique est tardive, voire absente, malgré la présence de lésions anatomiques encéphaliques parfois sévères. Par ailleurs, à coté des lésions de porencéphalie typique, l’imagerie cérébrale révèle diverses lésions de la substance blanche, dont le trait commun est la leucoencéphalopathie, celle-ci étant associée de façon variable à des infarctus cérébraux et à des microsaignements [17, 18].
Des progrès significatifs ont ensuite été réalisés dans la compréhension des mécanismes des atteintes neurologiques associées aux mutations de COL4A1 . Celles-ci semblent expliquées par l’existence d’une microangiopathie cérébrale. En effet, l’équipe de Gould a montré l’effet délétère de traumatismes à la naissance dans la survenue des hémorragies intracérébrales chez les animaux hétérozygotes présentant une délétion de l’exon 40 du gène, ce qui suggère une fragilité accrue des petits vaisseaux cérébraux à rapprocher d’anomalies de la membrane basale au microscope électronique [19]. Dans le même article, écrit en collaboration avec le groupe français d’Elisabeth Tournier-Lasserve et de Marie-Germaine Bousser, le phénotype d’une nouvelle famille, porteuse d’une substitution glycine à l’état hétérozygote dans l’exon 25 est rapporté. Contrairement aux descriptions antérieures, aucun patient ne présente de cavité porencéphalique. Les anomalies cérébrales associent une leucoencéphalopathie et la survenue d’hémorragies cérébrales d’expression clinique et de sévérité variables, en l’absence d’hypertension. Les traumatismes ou la prescription d’anticoagulants apparaissent comme des facteurs aggravants [19, 20].
De plus, des tortuosités artériolaires rétiniennes bilatérales sont pour la première fois rapportées dans cette même famille, élargissant ainsi le spectre des anomalies vasculaires associées aux mutations de COL4A1 [19]. Des anomalies similaires sont observées chez les souris dans certains fonds génétiques [16, 19, 21].
Dans les deux dernières années (2006 et 2007), les manifestations neurologiques et oculaires observées chez les patients avec des mutations du gène COL4A1 se sont encore enrichies. Un cas sporadique d’hémorragies intracérébrales récidivantes provoquées par des activités sportives à partir de l’âge de dix sept ans, en l’absence d’hypertension artérielle, a été rapporté [22]. Une mutation du gène COL4A1 a également été identifiée dans une famille présentant, outre une vasculopathie céré- brale, diverses anomalies du segment antérieur de l’œil (cataracte et glaucome congénitaux, amblyopie, hypoplasie iridienne, microcornée) caractéristiques du syndrome d’Axenfeld-Rieger [23]. Ces mêmes anomalies ophtalmologiques sont observées chez les souris présentant une délétion de l’exon 40 de Col4a1 ainsi que chez des souris
Raw , Bru et Svc , présentant chacune une mutation ponctuelle de
Col4a1 provoquée par mutagénèse aléatoire [24].
SYNDROME AHNAC : UNE NOUVELLE « BASALOPATHIE » SYSTÉ- MIQUE
Le réseau α1α1α2(IV) étant largement exprimé dans toutes les MB de l’organisme, il était prévisible que des mutations de COL4A1 pourraient se traduire par une atteinte systémique, non strictement limitée aux petits vaisseaux cérébraux ou rétiniens. La démonstration d’une atteinte diffuse des MB a d’abord été apportée par l’étude de trois nouvelles souches de souris, Raw , Bru et Svc , [24]. Le phénotype prédominant chez ces animaux est ophtalmologique, associant des anomalies de la cornée, du cristallin ou de l’iris, à un certain degré de rigidité des artérioles rétiniennes. L’étude ultrastructurale de plusieurs tissus, incluant l’œil, la peau, l’œsophage et l’aorte, a montré des anomalies des MB dont l’épaisseur est réduite avec présence d’interruptions focales. Dans le rein, les lésions sont strictement localisées à la capsule de Bowman, où une hyperplasie des cellules épithéliales pariétales s’associe à un plissement exagéré de la MB. Ces anomalies rénales sont exclusivement observées dans la lignée Bru , n’ont pas de traduction fonctionnelle, et chez ces animaux, la MBG et les MB tubulaires apparaissent normales [24].
Nous avons récemment rapporté trois familles dans lesquelles des mutations hétérozygotes de COL4A1 produisent un phénotype systémique associant des anomalies rénales, musculaires et vasculaires, touchant les vaisseaux de petit calibre et de gros calibre [1]. Les trois mutations produisant ce phénotype affectent toutes une glycine, respectivement en position G498, G519 et G529 (exons 24 et 25), dans une région très restreinte du domaine collagénique [1], (Figure 2). Les arbres généalogiques des trois familles ainsi que les mutations correspondantes du gène COL4A1 sont présentés dans la Figure 3 .
Manifestations rénales
Les signes rénaux sont caractérisés dans la première famille (mutation G498) par l’existence d’une hématurie glomérulaire chez tous les patients atteints. Alors que l’hématurie reste strictement isolée (sans protéinurie, ni hypertension artérielle, ni insuffisance rénale) chez les six patients des deux dernières générations, une insuffisance rénale terminale a émaillé l’histoire de deux ancêtres atteints [25]. Toutefois, la présence de facteurs aggravants, en particulier un état septique aigu chez l’un des patients, ne permet pas de rapporter de manière indiscutable cette évolution délétère à l’anomalie génétique.
Le phénotype rénal associé aux mutations G519 et G528 est caractérisé par la présence de volumineux kystes rénaux bilatéraux, asymptomatiques, siégeant à la fois dans le cortex et dans la médullaire (Figure 4A), et associés à une insuffisance rénale débutante, sans hypertension artérielle, ni hématurie ni protéinurie. La taille des kystes s’accroît avec l’âge, et un volumineux kyste hépatique est en outre présent chez l’un des patients. L’étude histologique du rein n’a pu être réalisée dans ces deux familles en raison de la pathologie kystique. Seuls quelques sujets porteurs de la mutation G498 présentent des kystes rénaux de petite taille et en faible nombre ; leur signification pathologique reste discutable.
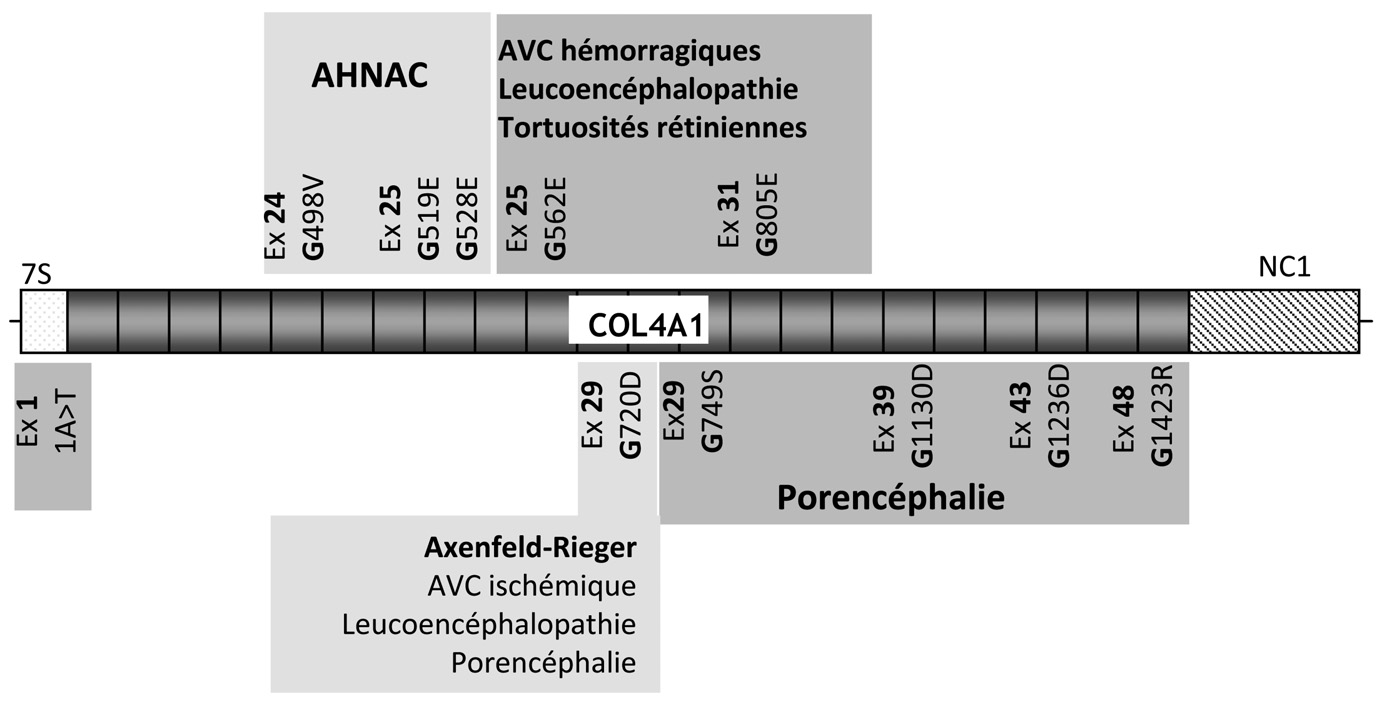

Fig. 2. — Mutations du gène
COL4A1 et phénotypes associés chez l’homme. Les différentes pathologies sont représentées par des couleurs différentes. Ex signifie exon, les 10 mutations présentes dans les séquences traduites remplacent une glycine (G) par un autre acide aminé représenté par une autre lettre;ˆ le nombre entre les deux lettres correspond à la position de la mutation dans la séquence d’acides aminés.
Fig. 3. — Arbres généalogiques des familles présentant le syndrome HANAC, et mutations correspondantes du gène COL4A1 . Les sujets malades dans chacune des familles sont identifiés par un symbole plein.
Fig. 4. — Anomalies rénales, vasculaires et des membranes basales au cours du syndrome AHNAC.
(A) Kystes rénaux bilatéraux de grande taille. (B) IRM cérébrale, montrant une leucoencéphalopathie bilatérale, cliniquement asymptomatique. (C) Angiographie rétinienne montrant un aspect typique de tortuosités artériolaires rétiniennes. (D) Angiographie cérébrale montrant trois formations anévrismales du siphon carotidien droit. (E, F) Microscopie électronique du rein montrant un épaississement de la MB tubulaire avec multilamination de la lamina densa (E) et des zones d’interruption focale de la MB d’un capillaire péritubulaire (F). (G) Microscopie électronique de la peau montrant des dédoublements multiples de la lame basale à la jonction dermoépidermique.
Manifestations vasculaires
Dans les trois familles, on retrouve une atteinte microvasculaire cérébrale qui se caractérise par la présence d’une leucoencéphalopathie (Figure 4B). Toutefois, elle est inconstante et n’a été symptomatique que chez un patient sous forme d’un accident ischémique transitoire cérébelleux. Tous les patients atteints présentent en outre des tortuosités artériolaires rétiniennes bilatérales typiques (Figure 4C).
Celles-ci sont asymptomatiques, ou se compliquent d’épisodes d’hémorragies rétiniennes responsables d’une baisse transitoire de l’acuité visuelle, se résolvant secondairement sans séquelle visuelle ni rétinienne. Des manifestations microvasculaires extracérébrales, en particulier un syndrome de Raynaud typique, sont notées chez plusieurs patients, alors qu’elles étaient apparemment absentes dans les familles précédemment publiées. De plus, l’étude angiographique cérébrale a permis l’identification dans les trois familles, de lésions anévrismales artérielles cérébrales, parfois multiples, se localisant électivement au niveau de la carotide interne droite ou dans un cas, au niveau de l’artère cérébrale moyenne (Figure 4D). A ce jour, aucune de ces lésions ne s’est compliquée de rupture. Ainsi, COL4A1 doit maintenant être considéré comme l’un des gènes potentiellement responsables d’une forme familiale dominante d’anévrismes intracérébraux.
Manifestations musculaires
La survenue de crampes est l’un des signes importants du syndrome AHNAC. Elles sont symptomatiques dès l’enfance, et sont spontanées ou déclenchées par certains mouvements, l’effort ou l’alcool. La force et la trophicité musculaires restent pré- servées. Les crampes s’associent à une élévation permanente des enzymes musculaires (CPK), qui dans l’une des familles, représente le seul signe d’atteinte musculaire (Famille 2, G519). La biopsie musculaire réalisée chez l’un des patients n’a pas montré d’anomalie significative des fibres musculaires striées, ni des vaisseaux adjacents, ni même des MB analysées en microscopie électronique.
La physiopathologie des manifestations musculaires reste hypothétique : elles pourraient résulter de phénomènes ischémiques transitoires, de microhémorragies à l’occasion d’effort musculaire particulier, ou encore d’un défaut d’interaction entre les fibres musculaires striées et la MB dans laquelle le trimère α1α1α2(IV) coexiste avec le collagène VI. Certaines myopathies héréditaires, parfois sévères, résultent en effet de mutations de gènes codant pour des constituants de la MB musculaire, comme le collagène VI (Myopathie de Bethlem) ou la chaîne α2 de la laminine (Dystrophie musculaire congénitale de type 1A).
Anomalies des membranes basales
L’étude ultrastructurale des MB du rein et de la peau, effectuée dans les familles 1, 2 et 3, a été très informative. Alors que la structure de la MBG est normale, les MB tubulaires et la MB de la capsule de Bowman sont irrégulières et épaissies de façon focale (Figure 4E). Dans les zones épaissies, la lamina densa apparaît inhomogène et feuilletée, et contient des logettes transparentes aux électrons. Ces aspects rappellent ceux observés dans la MBG au cours du syndrome d’Alport. Les MB des capillaires péritubulaires présentent elles aussi une paroi d’épaisseur parfois irrégulière, voire dédoublée ou multilamellaire, ainsi que des zones focales de discontinuité (Figure 4F). L’expression des chaînes α1 et α2 dans les diverses MB rénales apparaît néanmoins préservée en immunomicroscopie électronique.
Alors qu’il n’existe pas d’anomalie cutanée clinique, la jonction dermo-épidermique est anormale avec présence de zones de dédoublement ou de réplication pathologique de la lamina densa (Figure 4G). Dans les vaisseaux dermiques, il existe un épaississement irrégulier des lames basales entourant les cellules musculaires lisses de la média, qui apparaissent dissociées. Ces anomalies concourent vraisemblablement à la fragilité des parois vasculaires observée chez ces patients, et aux anomalies de la réactivité vasculaire chez les souris (Van Agtmael, communication personnelle).
MUTATIONS DU GÈNE
COL4A1 ET CORRÉLATIONS PHÉNOTYPE-
GÉNOTYPE
Bien que le nombre de familles avec une mutation identifiée du gène
COL4A1 soit encore très restreint, une grande diversité se dégage déjà de l’étude phénotypique, comme cela a été observé chez la souris. Dix des onze mutations actuellement connues sont responsables de la substitution d’un résidu glycine dans le domaine collagénique (Figure 2). Alors que les mutations à l’origine d’une porencéphalie sont localisées au delà de l’exon 25, à l’exception de la mutation du codon ATG, les trois mutations associées au syndrome AHNAC sont localisées dans une région de 90 nucléotides dans les exons 24 et 25. Une quatrième famille présente une mutation glycine dans la même région (Plaisier, communication personnelle). Plusieurs hypothèses peuvent être avancées pour expliquer l’effet pathogène différent de ces mutations : modification de l’interaction de la molécule de collagène IV mutée avec les autres constituants de la MB ou les cellules adjacentes ; augmentation variable de la sensibilité aux protéases de la protéine mutée ; modifications conformationnelles de la triple hélice d’importance variable en fonction de la localisation de la mutation glycine.
En outre, l’existence d’une variabilité phénotypique intrafamiliale dans les familles avec porencéphalie suggère une modulation possible par des gènes modificateurs et/ou des facteurs environnementaux. Ces deux points sont bien illustrés chez l’animal. En effet, le phénotype vasculaire rétinien n’est observé que lorsque la délétion de l’exon 40 de Col4a1 est exprimée chez la souris dans un fond génétique
C57Bl/6 [19]. L’influence du contexte génétique vaut aussi pour la dysgénésie du segment antérieur de l’œil et l’hypoplasie du nerf optique également observées chez la souris [21]. Un locus modificateur dominant pour ces manifestations aurait été identifié par l’équipe de Gould [21]. De plus, la survenue des hémorragies cérébrales néonatales est prévenue chez l’animal par la réalisation d’une césarienne, évitant ainsi le traumatisme de la naissance [19].
MUTATIONS DU GÈNE COL4A1 : QUELLES IMPLICATIONS POUR LE
DIAGNOSTIC ?
Hémorragies intracérébrales profondes d’étiologie indéterminée
En raison de l’absence de symptômes neurologiques pathognomomiques, d’anomalies spécifiques en IRM, et de la possibilité de porteurs asymptomatiques et de cas sporadiques, un certain nombre de patients avec une mutation du gène COL4A1 ne sont probablement pas identifiés. Une enquête génétique devrait être entreprise devant l’un au moins des arguments suivants, en particulier chez les jeunes adultes :
histoire personnelle ou familiale d’hémiparésie infantile ou de porencéphalie congé- nitale, leucoencéphalopathie avec microsaignements asymptomatiques en l’absence d’hypertension artérielle, présence de tortuosités des artérioles rétiniennes. La détection d’une mutation du gène COL4A1 a des implications importantes pour les femmes porteuses de la mutation chez lesquelles il faut conseiller un accouchement par césarienne, et pour tous les patients à risque chez lesquels il faut éviter les activités physiques intenses, les traumatismes, et surveiller très soigneusement les traitements anticoagulants [22].
Hématuries familiales et maladies kystiques rénales
Le phénotype rénal des familles ayant un syndrome AHNAC est une incitation à rechercher des mutations de COL4A1 au cours de formes autosomiques dominantes d’hématurie ou de maladies kystiques ne présentant pas les caractéristiques habituelles de polykystose autosomique dominante. Ces deux types de maladies doivent conduire à la recherche de signes extra-rénaux atypiques chez tous les membres de la famille, présentant ou non l’atteinte rénale : en particulier anomalies vasculaires rétiniennes grâce à l’examen du fond d’œil, anomalies musculaires ou simple élévation des CPK, voire anomalies cérébrales. Savige et col [26] ont testé, sans succès, l’implication du locus COL4A1/COL4A2 dans 15 familles d’hématurie familiale bénigne non liée au locus
COL4A3/COL4A4. Une étude de COL4A1 par analyse de polymorphysme de conformation simple brin ou SSCP (« Single Strand Conformation Polymorphism ») s’est elle aussi révélée négative chez 23 individus ayant les critères clinico-biologiques et ultrastructuraux de maladie des membranes basales fines [26]. Ces résultats n’excluent pas la responsabilité de mutations de COL4A1 dans certaines formes familiales d’hématurie, surtout si elles sont associées à des anomalies vasculaires rétiniennes.
CONCLUSION
Des mutations du gène
COL4A1 peuvent être responsables d’une « basalopathie » systémique, le syndrome AHNAC. Ce diagnostic devrait être évoqué dans les familles avec hématurie, maladie kystique des reins, anévrismes intracrâniens, et crampes musculaires à transmission autosomique dominante, surtout si ces manifestations s’accompagnent de tortuosités des artérioles rétiniennes.
L’étude phénotypique des patients ayant une mutation hétérozygote de COL4A1 apporte des éclairages importants sur le rôle du collagène IV et plus généralement des membranes basales en pathologie. En particulier, les anomalies microvasculaires et macrovasculaires attestent du rôle central des MB comme structure de soutien et de régulation du tonus vasculaire. L’établissement de modèles animaux du syndrome AHNAC pourra permettre de mieux comprendre la physiopathologie des anomalies observées.
REMERCIEMENTS
Nous souhaitons remercier toutes les personnes ayant contribué à la caractérisation du syndrome AHNAC : S. Alamowitch, C. Antignac, Y.S. Cohen, C. Combe, T. Desmettre, M. Dracon, M.
Fardeau, O. Gribouval, D. Kerjaschki, B. Marro, B. Mougenot, C. Prost, T. Van Agtmael, M.C.
Verpont.
BIBLIOGRAPHIE [1] Plaisier E., Gribouval O., Alamowitch S. et al — Role of COL4A1 Mutations in the
Hereditary Angiopathy with Nephropathy, Aneurysm and Cramps (HANAC) Syndrome.
New Engl. J. Med. , 2007, 357 , 31-9.
[2] Kalluri R. — Basement membranes : structure, assembly and role in tumour angiogenesis.
Nature Cancer Rev. , 2003, 3 , 422-433.
[3] Miner J.H., Sanes J.R. — Collagen IV alpha 3, alpha 4, and alpha 5 chains in rodent basal laminae : sequence, distribution, association with laminins, and developmental switches. J. Cell Biol. , 1994, 127 , 879-891.
[4] Hudson B.G., Tryggvason K., Sundaramoorthy M. et al. — Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen.
N. Engl. J. Med., 2003, 348 , 2543-2556.
[5] Sundaramoothy M., Meiyappan M., Todd P. et al — Crystal structure of NC1 domains :
structure basis of type IV collagen assembly in basement membranes. J. Biol. Chem., 2002, 277 , 31142-31153.
[6] Barker D.F., Hostikka S.L., Zhou J. et al — Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science , 1990, 248 , 1224-1227.
[7] Mochizuki T., Lemmink H.H., Mariyama M. et al — Identification of mutations in the alpha3(IV) and alpha4(IV) collagen genes in autosomal recessive Alport syndrome.
Nat. Genet , 1994, 8 , 77-81.
[8] Lemmink H.H., Nillensen W.N., Mochizuki T. et al — Benign familial hematuria due to mutation of the type IV collagen alpha4 gene.
J. Clin. Invest. , 1996, 98 , 1114-1118.
[9] Miner J.H. — Renal basement membrane components.
Kidney Int , 1999, 56 , 2016-2024.
[10] Kalluri R., Shield C.F., Todd P. et al — Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to proteolysis. J. Clin. Invest. , 1997, 99 , 2470-2478.
[11] Zeisberg M., Khurana M., Rao V.H. et al — Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease.
PLoS Med. , 2006, 3 , e100.
[12] Jais J.P., Knebelmann B., Giatras I. et al — X-linked Alport syndrome : natural history in 195 families and genotype- phenotype correlations in males.
J. Am. Soc. Nephrol. , 2000, 11 , 649-657.
[13] Gross O., Netzer K.O., Lambrecht et al. — Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome : impact on clinical counselling.
Nephrol Dial Transplant , 2002, 17 , 1218-1227.
[14] Jais J.P., Knebelmann B., Giatras I. et al — X-linked Alport syndrome : natural history and genotype-phenotype correlations in girls and women belonging to 195 families : a ‘‘ European Community Alport Syndrome Concerted Action ’’ study. J. Am. Soc. Nephrol. , 2003, 14, 2603-2610.
[15] Poschl E., Schlotzer-Schrehardt U., Brachvogel B. et al — Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development , 2004, 131 , 1619-1628.
[16] Gould D.B., Phalan F.C., Breedveld G.J. et al — Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science , 2005, 308 , 1167-1171.
[17] Breedveld G., De Coo R.F., Lequin M.H. et al — Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly.
J. Med. Genet. , 2006, 43 , 490-495.
[18] Van der Knaap M.S., Smit L.M., Barkhof F. et al — Neonatal porencephaly and adult stroke related to mutations in collagen IV A1.
Ann. Neurol. , 2006, 59 , 504-511.
[19] Gould D.B., Phalan F.C., Van Mil S.E. et al — Role of COL4A1 in small-vessel disease and hemorrhagic stroke.
N. Engl. J. Med. , 2006, 354 , 1489-1496.
[20] Vahedi K., Massin P., Guichard J.P. et al — Hereditary infantile hemiparesis, retinal arteriolar tortuosity, and leukoencephalopathy.
Neurology , 2003, 60 , 57-63.
[21] Gould D.B., Marchant J.K., Savinova O.V., Smith R.S., John S.W. — Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum. Mol.
Genet., 2007, 16 , 798-807.
[22] Vahedi K., Boukobza M., Massin P., Gould D.B., Tournier-Lasserve E., Bousser M.G. — Clinical and brain MRI follow-up study of a family with COL4A1 mutation. Neurology, 2007, 69 , 1564-1568.
[23] Sibon I., Coupry I., Menegon P. et al — COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke.
Ann. Neurol., 2007, 62, 177-184.
[24] Van Agtmael T., Schlotzer-Schrehardt U., McKie L. et al. — Dominant mutations of
Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum. Mol. Genet. , 2005, 14 , 3161-3168.
[25] Plaisier E., Alamowitch S., Gribouval O. et al — Autosomal-dominant familial hematuria with retinal arteriolar tortuosity and contractures : a novel syndrome . Kidney Int. , 2005, 67 , 2354-2360.
[26] Savige J., Zhang K.W., Wang Y.Y. et al — Examination of the COL4A1 gene as a further locus for thin basement membrane nephropathy.
J. Am. Soc. Nephrol. , 2005, 16 , 367A (abstract).
DISCUSSION
M. Pierre GODEAU
Y a-t-il un rapport entre cette maladie et la maladie des membranes basales minces (ou fines) qui s’accompagne d’hématurie microscopique ?
La maladie des membranes basales minces se manifeste par une hématurie familiale à transmission autosomique dominante. Cette hématurie est d’origine glomérulaire et est associée dans les cas typiques, à un amincissement de la membrane basale glomérulaire au microscope électronique. Par contre, il n’existe pas d’anomalie des membranes basales extraglomérulaires, l’adjectif « mince » ne qualifiant que les membranes basales glomé- rulaires. Cette pathologie entre dans le cadre des hématuries familiales dites bénignes dont 50 % environ sont associés à des mutations des chaînes α3 et α4 du collagène de type IV (gènes COL4A3 et COL4A4 ). Au contraire de la maladie des membranes basales minces, les membranes basales glomérulaires sont strictement normales dans le syndrome AHNAC qui est associé à des mutations du gène COL4A1 . Cela n’est pas surprenant si l’on considère que la chaîne α1 du collagène de type IV n’est que très faiblement représentée dans la membrane basale glomérulaire mature (alors qu’elle est prédominante au sein du trimère α1α2α2(IV) dans les phases précoces de l’embryogénèse glomérulaire). L’origine de l’hématurie dans le syndrome AHNAC n’est vraisemblablement pas glomérulaire. Les anomalies ultrastructurales majeures des membranes basales des tubules rénaux et des capillaires interstitiels du rein suggèrent que l’hématurie pourrait être vasculaire et tubulaire.
M. Gabriel RICHET
Avez-vous recherché la myoglobinurie qui pourrait être associée aux crampes musculaires ?
Nous n’avons pas trouvé de myoglobinurie associée aux crampes musculaires. La seule anomalie constatée était une élévation de la créatinine-phospho-kinase (CPK). Nous n’avons pas observé de dégradation de la fonction rénale contemporaine des épisodes d’hématurie, qui souvent accompagne la myoglobinurie.
M. Christian NEZELOF
Avez-vous pu contrôler l’état du placenta qui est une éponge vasculaire ? À côté des cellules endothéliales et épithéliales existe-t-il d’autres cellules capables d’exprimer cette anomalie génétique ?
Nous n’avons pas pu obtenir de placenta pour l’analyse des lésions vasculaires. Le placenta est vraisemblablement un organe affecté par le syndrome AHNAC, comptetenu de sa très riche vascularisation. Les fibroblastes cutanés produisent également la chaîne α1 du collagène de type IV. Nous les utilisons en culture primaire pour établir le diagnostic génétique de la maladie à partir de l’ARN rétrotranscrit, et pour comprendre les anomalies de sécrétion et de trafic de la chaîne mutée.
M. Jean-Daniel SRAER
Y a-t-il dans ces cas là une insuffisance rénale puisque c’est une maladie systémique avec atteinte vasculaire ?
Dans la première famille où le syndrome s’est manifesté par une hématurie, plusieurs patients ont présenté une insuffisance rénale dans un contexte pathologique (thrombose cave, sepsis) qui rend difficile l’interprétation de cette insuffisance rénale. En revanche, dans les deuxième et troisième familles, les kystes sont associés à une insuffisance rénale de degré modérée (débit de filtration glomérulaire entre 40 et 50 ml/min/1,73m2) qui n’est pas expliquée par le volume des kystes. Cette insuffisance rénale est vraisemblablement liée à une atteinte du parenchyme dont l’origine n’a pu être établie en raison de l’impossibilité d’effectuer une biopsie rénale. Par ailleurs, compte-tenu du rôle important de COL4A1 aux étapes précoce de la formation des glomérules, il n’est pas exclu que ces patients présentent une réduction néphronique congénitale.
M. Raymond ARDAILLOU
Y a-t-il des anomalies de la voie de signalisation Notch dans le syndrome AHNAC qui réunit kystes rénaux et anomalies artériolaires ?
Nous n’avons pas exploré la voie de signalisation Notch dans le syndrome AHNAC.
Certaines caractéristiques de la maladie (atteinte de la paroi vasculaire avec dissociation des cellules musculaires lisses, lésions de la substance blanche d’origine ischémique vraisemblable en l’absence d’hypertension artérielle) rappellent le syndrome CADASIL dans lequel il existe des mutations du gène Notch 3 .
M. Jean-Yves LE GALL
Quelle est l’évolution naturelle de la maladie ? A-t-elle fait l’objet de diagnostics anténataux ?
La maladie semble relativement bénigne. Nous n’avons pas observé d’accidents ischémiques sévères comme dans les formes cérébrales de la maladie associées aux mutations de COL4A1 décrites antérieurement. Toutefois, nous ne connaissons pas le risque hémorragique des anévrysmes intracraniens, et l’un des membres de la première famille a présenté une hémorragie cérébrale traumatique. La prudence incite à éviter les traumatismes (sports violents) dans ce syndrome, et à discuter avec une particulière attention l’indication des traitements anticoagulants ou anti-agrégants plaquettaires. La maladie n’a pas fait l’objet de diagnostics anténataux.
M. Pierre RONDOT
Attribuez-vous les lésions de la substance blanche cérébrale à l’atteinte vasculaire, ce qui n’est pas le cas dans les ischémies cérébrales ? N’y a-t-il pas d’atteinte de glyco ou de lipoprotéines cérébrales ?
Nous n’avons pas étudié les glycoprotéines et les lipoprotéines cérébrales. Toutefois, les anomalies ultrastructurales de la paroi vasculaire, le phénomène de Raynaud observé chez certains membres atteints, l’absence d’hypertension artérielle sont en faveur d’une origine ischémique des lésions de la substance blanche. Des micro-hémorragies « blebs » ont également été mises en évidence.
M. Charles MENKÈS
L’augmentation des CPK est-elle constante et correlée avec l’importance et la diffusion des crampes ?
L’augmentation des CPK est constante chez les sujets porteurs d’une mutation de COL4A1 mais apparemment non corrélée à l’intensité et la diffusion des crampes.
Bull. Acad. Natle Méd., 2008, 192, no 5, 971-986, séance du 13 mai 2008