
Résumé
La transition épithélio-mésenchymateuse (TEM) est un processus morphogénique fondamental au cours duquel les cellules perdent un nombre important de leurs caractéristiques épithéliales pour acquérir des propriétés de cellules mésenchymateuses. Les mécanismes mis en jeu sont fréquemment réactivés au cours de la progression tumorale, générant des cellules douées de capacités de motilité et d’invasion cellulaire. Ainsi, de nombreuses observations in vitro et in vivo tendent aujourd’hui à démontrer le rôle de la TEM dans le processus de dissémination métastatique des tumeurs d’origine épithéliale. Récemment, différents facteurs de transcription impliqués dans l’induction de la TEM, en particulier les protéines Twist, ont été montrés comme étant capables d’inhiber les réponses onco-suppressives normalement mises en œuvre pour contrecarrer l’émergence d’une population cellulaire présentant une activité mitogénique aberrante. L’échappement à ces processus de sauvegarde, qui conduisent normalement à une apoptose ou à une sénescence prématurée des cellules hyperprolifératives, constitue une étape essentielle de l’évolution de la pathologie cancéreuse d’un stade bénin à un stade malin. Ces observations suggèrent que la TEM pourrait jouer un rôle crucial tant dans le développement de la tumeur primaire que de celui de la dissémination métastatique. Dans cette revue, nous présenterons les principales données sur lesquelles repose cette hypothèse et nous discuterons de leurs possibles implications cliniques.
Summary
During epithelial-mesenchymal transition (EMT) — a morphogenetic program involved in several steps of embryogenesis — epithelial cells lose many of their epithelial characteristics and acquire properties typical of mesenchymal cells. The mechanisms underlying this transition are frequently reactivated during tumor progression, generating cells with enhanced motility and invasiveness. Several in vitro and in vivo studies point to a role of EMT in metastatic dissemination of epithelial tumors. In addition, recent data show that EMTinducing transcription factors, such as the Twist proteins, can inhibit crucial oncosuppressive responses that normally counteract the emergence of a cell population with aberrant mitogenic activity, by triggering either apoptosis or premature senescence. Abrogation of these failsafe cellular mechanisms is a prerequisite for malignant progression. Together, these observations suggest that EMT could play a major pathological role by favoring both tumor development and metastatic dissemination. We examine the main data supporting this hypothesis and discuss its potential clinical implications.
INTRODUCTION
La transformation maligne et la croissance d’une tumeur primaire sont dictées par l’accumulation d’altérations génétiques et épigénétiques qui vont conférer progressivement aux cellules des capacités accrues de division, une insensibilité aux signaux inhibiteurs de prolifération et un échappement au processus de vieillissement cellulaire (sénescence réplicative) et à la mort cellulaire programmée (apoptose) [1].
Après une phase initiale de développement tumoral, les cellules cancéreuses (ou pré-cancéreuses) se retrouvent rapidement dans des conditions d’hypoxie à l’origine de l’induction d’un programme de néo-angiogenèse. On estime que ce développement de nouveaux vaisseaux est nécessaire à la progression tumorale dès que la tumeur atteint une taille de 1 mm [2]. Les tumeurs d’origine épithéliale sont les tumeurs les plus fréquentes de l’adulte. Au cours des phases initiales du processus malin, les cellules tumorales restent confinées au site initial du fait de l’importance des jonctions inter-cellulaires (jonctions serrées, jonctions adhérentes ou desmosomes) qui caractérisent les tissus épithéliaux. Les cellules épithéliales normales présentent une polarisation apico-basale, une répartition localisée des molécules d’adhésion (cadhérines, intégrines), une latéralisation des jonctions inter-cellulaires et une polarisation des fibres d’actine. En outre, elles sont ancrées par leur pôle basal à un réseau dense de glycoprotéines et de protéoglycanes, appelé membrane basale, qui délimite le tissu épithélial de la matrice extracellulaire environnante. Cependant, au cours de la progression tumorale, les lésions peuvent devenir invasives par perte des jonctions cellulaires et franchissement de la membrane basale. Par une succession d’étapes, appelée « cascade métastatique », les cellules envahissent alors la matrice extracellulaire, pénètrent dans les vaisseaux sanguins ou lymphatiques (processus d’intravasation), circulent dans le flux sanguin ou lymphatique, se logent dans des microvaisseaux d’un tissu à distance de la tumeur d’origine, envahissent le parenchyme de celui-ci (processus d’extravasation) pour former des micrométastases, certaines de celles-ci pouvant à terme évoluer pour former des métastases [3].
Cette cascade métastatique correspond donc à une succession d’étapes, le passage de l’une à l’autre nécessitant l’acquisition par les cellules tumorales d’un nombre significatif de propriétés intrinsèques particulières. Ces événements incluent notam- ment la perte des jonctions inter-cellulaires et la production de métalloprotéases pour l’étape d’invasion, l’acquisition de capacités de survie dans la circulation, et le gain de capacités d’adaptation dans un nouveau microenvironnement. Le franchissement de ces étapes repose également sur le développement d’interactions entre cellules tumorales et cellules du microenvironnement, conduisant, en particulier, à la production de molécules pro-angiogéniques par les cellules endothéliales, les cellules du stroma et certaines cellules progénitrices du système hématopoïétique, et à la sécrétion de métalloprotéases, de cathepsines et de glycosidases par les cellules stromales [4].
Selon le modèle classique de la progression tumorale, la croissance d’une tumeur évolue selon des phases d’expansion clonale qui font suite à la sélection de cellules ayant acquis des avantages de prolifération et de survie [5]. La pression de sélection joue également un rôle probable dans la colonisation du tissu hôte et la formation de macrométastases. À ce titre, la nécessité pour les cellules tumorales d’acquérir des propriétés leur permettant de s’adapter à leur nouvel environnement apporte une explication partielle au délai important, correspondant au processus de « dormance micrométastatique », constaté entre leur arrivée au site secondaire et le développement de la métastase [3, 6]. En revanche, il est plus difficile d’appréhender le rôle de la pression de sélection au cours des autres étapes de la cascade métastatique, en particulier au cours de l’invasion, de l’intravasation et de l’extravasation. Comment expliquer en effet la sélection dans la tumeur primaire de cellules ayant acquis des propriétés qui ne leur confèreront des avantages que dans les étapes ultérieures de la progression tumorale ? L’hypothèse que nous présentons ici repose sur la réactivation d’un programme transcriptionnel embryonnaire permettant aux cellules pré- cancéreuses et cancéreuses d’échapper à certaines contraintes onco-suppressives intrinsèques, et d’acquérir les propriétés de motilité, d’invasion et d’auto-renouvellement nécessaires à leur dissémination et à la génération de métastases.
Le processus de transition épithélio-mésenchymateuse et la dissémination métastatique
La transition épithélio-mésenchymateuse (TEM) est un processus dynamique complexe par lequel les cellules épithéliales se dédifférencient sous forme d’un phénotype cellulaire mésenchymateux plus mobile [7] (Figure 1). La TEM est caractérisée par une perte de la polarité cellulaire et une altération de l’organisation du cytosquelette et de l’adhésion cellulaire, permettant aux cellules d’acquérir une grande plasticité et une capacité à quitter le tissu épithélial pour migrer vers un site distant. Elle constitue un processus essentiel au cours de l’embryogenèse. En effet, en son absence, le développement de l’organisme ne dépasse pas le stade de la blastula. La TEM est ainsi nécessaire au développement du mésoderme au cours de la gastrulation, mais aussi à la migration des cellules de la crête neurale, puis à la formation des valves cardiaques et au développement du palais [8]. Elle est également observée au cours de la formation du placenta, et de la production de fibroblastes au cours de l’inflammation et de la cicatrisation. Plus récemment, un processus de trans-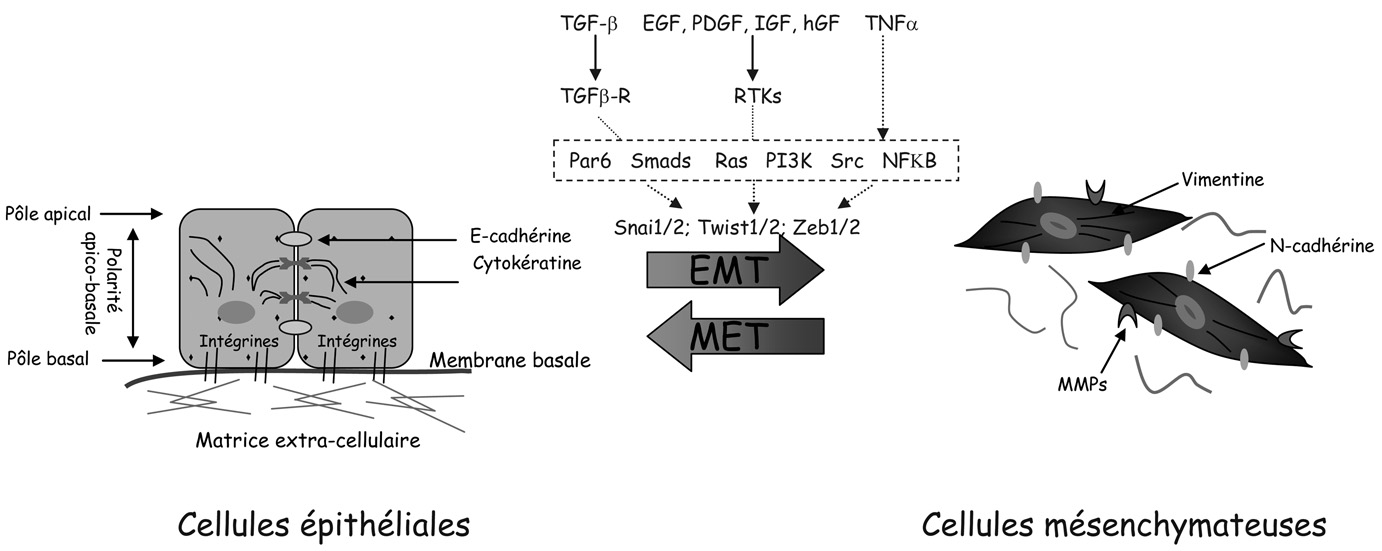
Fig. 1. — Processus de transition épithélio-mésenchymateuse (TEM) et de transition mésenchymoépithéliale (TME) Les cellules épithéliales différenciées sont des cellules polarisées, fortement associées les unes aux autres par des jonctions serrées et des jonctions adhérentes et reposant sur une membrane basale. La transition épithélio-mésenchymateuse (TEM) est un processus dynamique caracté- risé par la perte des jonctions inter-cellulaires, une profonde réorganisation du cytosquelette d’actine avec formation de fibres de stress et la conversion des filaments intermédiaires d’un type épithélial (cytokératine) à un type mésenchymateux (vimentine).
TGF-β: transforming growth factor-β ; EGF: epidermal growth factor, PDGF: platelet-derived growth factor ; IGF: insulin-like growth factor ; hGF: hepatocyte growth factor ; TNF-α:
tumor necrosis factor alpha ; RTKs: Récepteurs à activité Tyrosine Kinases ; TGFβ-R: Récepteur au TGF-β ; PI3K: Phosphoinositide 3-kinases ; Src: proto-oncogène Src ; NFKB: nuclear factor kappa-light-chain-enhancer of activated B cells.
différenciation faisant appel à des mécanismes similaires à ceux mis en jeu au cours de l’embryogenèse a été décrit dans deux processus pathologiques, la fibrose, en particulier la fibrose rénale, et les cancers [9, 10]. Il est à noter que le processus de transdifférenciation recouvre un large spectre de modifications inter- et intracellulaires, qui peuvent se produire de façon partielle. La TEM, en particulier dans un contexte pathologique, n’est donc pas nécessairement un changement complet de lignage cellulaire, les cellules résultantes pouvant présenter des caractéristiques de cellules mésenchymateuses et de cellules épithéliales.
La TEM est orchestrée par l’activation d’une machinerie transcriptionnelle complexe, mise en œuvre en réponse à des signaux du microenvironnement [7]. En culture cellulaire, la TEM peut être soit stable, c’est-à-dire que le phénotype mésenchymateux est maintenu en dépit de l’extinction du stimulus initiateur, ou transitoire, les cellules subissant une transition mésenchymo-épithéliale (TME) après disparition de la signalisation activatrice. Une grande variété de signaux extracellulaires peuvent induire une TEM [7, 10]. De façon intéressante, la plupart des voies impliquées au cours du processus embryonnaire sont également connues pour être activées au cours de la progression tumorale. C’est le cas des voies Wnt, Hedgehog ou Notch, ainsi que des voies de signalisation empruntées par les cytokines de types TGF-β ( transforming growth factor- β), EGF ( epidermal growth factor ),
FGF ( fibroblast growth factor ) et HGF ( hepatocyte growth factor) [7]. Le TGF-β est sans doute la cytokine dont le rôle au cours de la TEM est le mieux caractérisé [11].
La liaison à son récepteur transmembranaire, induit une hétérodimérisation de celui-ci et une activation qui conduit à la phosphorylation intra-cytoplasmique de protéines impliquées dans le contrôle de la polarité cellulaire et de la formation des jonctions serrées (protéine Par6), ainsi que des facteurs de transcription Smad2 et Smad 3. Suite à leur assemblage avec Smad4, le complexe des protéines Smad migre dans le noyau pour activer la transcription de gènes impliqués dans la TEM [12]. Le TGF-β active également des voies de signalisation indépendantes des protéines Smad, telles que la voie des MAP-kinases et des c-Jun-N terminal kinases, et celle des petites protéines de type GTP-ase (Rho, Rac et Cdc42), dont l’activation va provoquer une réorganisation du cytosquelette d’actine. Le TGF-β est enfin capable d’induire d’autres voies de signalisation impliquées dans l’induction d’une TEM, telles que les voies Notch et Wnt. A titre d’exemple, par l’intermédiaire de la voie PI3K/Akt, il provoque une inhibition de la glycogène synthase kinase-3β (GSK-3β).
Cette étape cruciale de l’activation de la voie Wnt conduit à la stabilisation et à la translocation dans le noyau de la β-caténine, où elle agira comme une sous-unité d’un complexe transcriptionnel capable d’induire l’expression de facteurs de transcription impliqués dans l’induction de la TEM (Snail1/Snail, Snail2/Slug, Twist1…) [13]. Différents facteurs de transcription sont impliqués dans les processus de TEM embryonnaire, qui agissent probablement de manière concertée. Certains appartiennent à la grande famille des facteurs de type bHLH (facteurs de transcription possédant un domaine basique hélice-boucle-hélice) tels que Twist1, Twist2 ou E47.
D’autres sont des facteurs de transcription à doigts de zinc (Snail1/Snail, Snail2/Slug, Zeb1, Zeb2/Sip1) [14]. Ces facteurs induisent une TEM en réprimant, de façon directe ou indirecte, la transcription de gènes codant pour des éléments clefs des jonctions adhérentes, telle que la E-cadhérine. De plus, certains de ces facteurs sont capables de réprimer l’expression d’autres protéines de jonctions cellulaires, d’activer l’expression de gènes mésenchymateux (N-cadhérine activée par Twist 1) et d’induire un remodelage complet du cytosquelette conduisant à une augmentation des capacités de migration.
L’hypoxie est un des mécanismes physiologiques capables d’induire une TEM au cours de la progression tumorale. Expérimentalement, des taux faibles d’oxygène provoquent une transdifférenciation de multiples lignées cellulaires d’origine épithé- liale [15]. Ce processus repose sur deux phases successives. La phase précoce est induite par une augmentation transitoire de la génération d’espèces réactives de l’oxygène et est caractérisée par l’inhibition de la GSK-3β et la translocation de Snail1, responsable de la répression de la transcription du gène de la E-cadhérine. La seconde phase, qui est accompagnée de l’acquisition des propriétés d’invasion, est caractérisée par l’induction du VEGF (vascular endothelial growth factor) et du facteur HIF-1α, inducteur transcriptionnel de Twist 1 [16].
L’observation d’une réactivation fréquente des facteurs embryonnaires Snail 1, Snail 2 et Twist 1 au cours de la progression tumorale constitue un argument important en faveur du rôle de la TEM au cours de la dissémination métastatique [17-19]. En concordance avec cette hypothèse, la comparaison du potentiel métastatique de lignées cellulaires mammaires chez la souris a permis de mettre en évidence une association étroite entre l’expression de Twist 1 et leur potentiel métastatique [20]. Expérimentalement, l’expression forcée dans des cellules épithé- liales humaines de Twist 1, de Snail1 ou de Snail2 (ou leur induction en réponse au VEGF ou à l’hypoxie) favorise la survenue d’une TEM, accompagnée de l’acquisition de propriétés migratoires et invasives et de l’augmentation de leur potentiel métastatique [20, 21]. À l’inverse, l’expression de la E-cadhérine, protéine clef des jonctions inter-cellulaires des tissus épithéliaux, est généralement perdue dans les stades invasifs et métastatiques des cancers du sein, dans les cancers du poumon non à petites cellules ayant envahi le ganglion drainant, ou dans les cancers colorectaux ayant métastasé dans le foie [22-24]. De façon remarquable, dans les cellules épithéliales mammaires, la seule perte de la E-cadhérine favorise l’apparition d’une TEM et augmente in vivo le potentiel métastatique de cellules transformées [25], démontrant le rôle essentiel joué par cette protéine.
En dépit des observations rapportées précédemment, la participation in vivo de la
TEM dans le processus d’invasion et de métastase a longtemps constitué un sujet important de controverse [26]. Deux arguments critiques principaux peuvent en effet être avancés. Les pathologistes ne relèvent jamais la présence de cellules en TEM au sein d’une tumeur épithéliale et les métastases provenant de carcinomes (tumeurs d’origine épithéliale) présentent un aspect épithélial et non mésenchymateux. La difficulté d’observer une cellule tumorale ayant subi une TEM au sein d’un échantillon clinique est probablement due au manque de méthodes spécifiques permettant de les différencier formellement d’une cellule mésenchymateuse d’origine stromale. Cet aspect est d’autant plus important que le processus de transdifférenciation est principalement sous la dépendance de signaux provenant du microenvironnement. Les cellules tumorales à la périphérie de la lésion, c’est-à-dire à proximité immédiate du stroma, sont de ce fait celles qui présentent la plus forte susceptibilité de subir une transdifférenciation. L’aspect épithélial des métastases peut être expliqué, quant à lui, par le caractère transitoire et réversible du processus.
Différents arguments expérimentaux suggèrent en effet que les cellules ayant subi une TEM recouvrent leur phénotype épithélial au cours des phases tardives de la dissémination métastatique par un processus de transition mésenchymo-épithéliale (TME) afin se s’adapter au mieux à leur nouveau micro-environnement. Une hypothèse alternative est que les cellules en TEM maintiennent leur phénotype mésenchymateux dans le site secondaire mais, par un processus de division asymé- trique, génèrent des cellules plus différenciées, de phénotype épithélial.
Malgré les difficultés technologiques d’identification des cellules en TEM, différents travaux ont apporté des arguments forts soutenant le rôle de ce processus au cours de la progression tumorale que ce soit dans des échantillons cliniques ou dans des modèles expérimentaux. À titre d’exemple, l’équipe de Brabletz a pu montrer, sur des échantillons de cancers colorectaux, que les cellules tumorales sur le front invasif présentaient des marqueurs de cellules dédifférenciées et des caractéristiques de cellules ayant subi une TEM (perte de la E-cadhérine, acquisition de marqueurs mésenchymateux) [27]. En lien avec une TEM, la membrane basale est rompue au niveau de ce front invasif, les cellules cancéreuses formant alors des bourgeons envahissant le stroma environnant. La membrane basale est à nouveau présente dans les métastases des ganglions lymphatiques et les métastases à distance, appuyant ainsi la notion de caractère transitoire de la TEM et l’existence probable d’un processus de transition mésenchymo-épithéliale (TME) dans le site secondaire.
De façon intéressante, la présence de cellules en TEM sur le front invasif de ces tumeurs est corrélée à un mauvais pronostic et à la présence de métastases à distance. Par ailleurs, l’équipe de Robert Weinberg a démontré, dans un modèle de xénogreffes de cellules épithéliales mammaires transformées, que les cellules présentes sur le front invasif des tumeurs exprimaient des marqueurs de cellules mésenchymateuses. Enfin, la démonstration formelle de l’existence du processus de TEM in vivo a été apportée par l’équipe de Leone grâce au développement d’un modèle de souris transgéniques présentant des cellules stromales ou des cellules épithéliales mammaires marquées irréversiblement, quel que soit leur devenir. Le suivi des cellules tumorales mammaires d’origine épithéliale a permis ainsi d’observer une TEM in vivo au cours de la carcinogenèse mammaire [28].
Au total, il existe aujourd’hui de nombreuses observations tant in vitro qu’ in vivo qui tendent à confirmer le rôle de la TEM au cours d’étapes clefs de la cascade métastatique. La perte des jonctions inter-cellulaires, la réorganisation du cytosquelette permettant la formation de prolongements cytoplasmiques et la contraction des cellules, et la production de protéases dégradant la matrice extracellulaire, donnent aux cellules tumorales la possibilité de se dissocier du tissu épithélial d’origine, d’envahir le stroma et les tissus environnants et facilitent les processus d’intravasation, puis d’extravasation.
Un rôle précoce de la TEM au cours de la progression tumorale ?
Au cours de ces dernières années, notre équipe a montré que, outre leur rôle dans le processus d’invasion et de dissémination métastatique, certains facteurs de transcription impliqués dans l’induction d’une TEM pouvaient également jouer un rôle oncogénique lors des phases précoces du développement de la tumeur. Ils inhibent en effet deux processus onco-suppresseurs essentiels, normalement mis en œuvre par les cellules en réponse à une activation mitogénique anormale : l’apoptose et la sénescence prématurée.
Les processus de sénescence et d’apoptose comme systèmes de sauvegarde cellulaire
L’homéostasie tissulaire est un équilibre très finement contrôlé pour se prémunir de l’apparition de cellules qui échapperaient à tout contrôle. Aussi, l’activation constitutive d’un oncogène dans une cellule normale ne procure aucun avantage sélectif aux cellules mais, à l’inverse, induit la mort cellulaire programmée (apoptose) ou l’arrêt définitif de la prolifération (sénescence), deux mécanismes de surveillance cellulaire nécessaires au maintien de l’intégrité du tissu et de l’organisme entier [29].
Un acteur clef de ces systèmes de surveillance cellulaire est la protéine p53, généralement présentée comme le « gardien de l’intégrité du génome ». Bien que d’aspect emphatique, cette formulation est probablement restrictive. En effet, l’activité de p53 n’est pas restreinte à la réponse cellulaire à des agents génotoxiques, qu’ils soient endogènes ou exogènes. Elle est également induite après des stress métaboliques ou oncogéniques, possiblement en l’absence de toute anomalie génétique [30]. Le type de réponse, apoptose ou sénescence, est dépendant du contexte micro-environnemental, du type cellulaire et de la nature de l’activation mitogénique aberrante (Figure 2). Ainsi, l’activation anormale des oncoprotéines Myc (c-Myc ou N-Myc) conduit essentiellement à un processus d’apoptose par activation de la protéine p14ARF qui neutralise la protéine HDM2 et empêche ainsi la dégradation de la protéine p53 [31, 32]. p53 ainsi stabilisée induit alors ses gènes cibles incluant des membres pro-apoptotiques de la famille Bcl2 comme Bax, Bim, Puma ou Noxa. À l’inverse, une activation anormale des oncoprotéines Ras ou ErbB2 induit préférentiellement une réponse de type sénescence prématurée, par induction des inhibiteurs des kinases dépendantes des cyclines (cdk) p21CIP1/WAF1 et p16INK4A [33]. Ces deux protéines maintiennent la protéine onco-suppressive Rb sous sa forme active hypophosphorylée, empêchant ainsi le facteur de transcription E2F d’induire l’expression des gènes nécessaires à la prolifération, aboutissant ainsi à un arrêt irréversible de la prolifération en phase G1 du cycle cellulaire. Les deux voies jouent des rôles distincts dans le processus de sénescence : l’induction de p21CIP1/WAF1, qui dépend de p53, est un processus précoce et entraîne un arrêt rapide du cycle cellulaire et une entrée en phase quiescente, alors que l’activation soutenue de la voie p16INK4A va figer la cellule dans un état irréversible, en inhibant à long terme l’expression de gènes cibles de E2F.
On estime aujourd’hui que le développement d’une tumeur primaire requiert deux grands types d’événements intrinsèques au cours des phases précoces du processus:
l’activation de voies permettant d’augmenter les capacités de prolifération cellulaire et l’inhibition de mécanismes onco-suppresseurs (sénescence et apoptose) qui sont normalement activés pour prévenir toute prolifération anormale. Ce modèle apporte une explication rationnelle au fait, qu’ in vitro , les oncogènes de types Myc ou Ras ne peuvent transformer des cellules qu’en l’absence de la protéine oncosuppressive p53. Il explique également la fréquence de l’inactivation de p53 dans les cellules cancéreuses [30]. Tous types confondus, plus de 50 % des cancers humains présentent en effet une perte de p53 par altération génétique (perte d’une copie du gène par délétion et altération de l’autre copie par mutation ponctuelle). En outre, différents mécanismes d’inactivation fonctionnelle de la protéine p53 ont été identifiés, en particulier dans certains cancers d’origine virale. De façon apparemment paradoxale, il existe cependant des cancers présentant une très forte activation oncogénique en l’absence d’altération du gène ou de la protéine onco-suppressive.
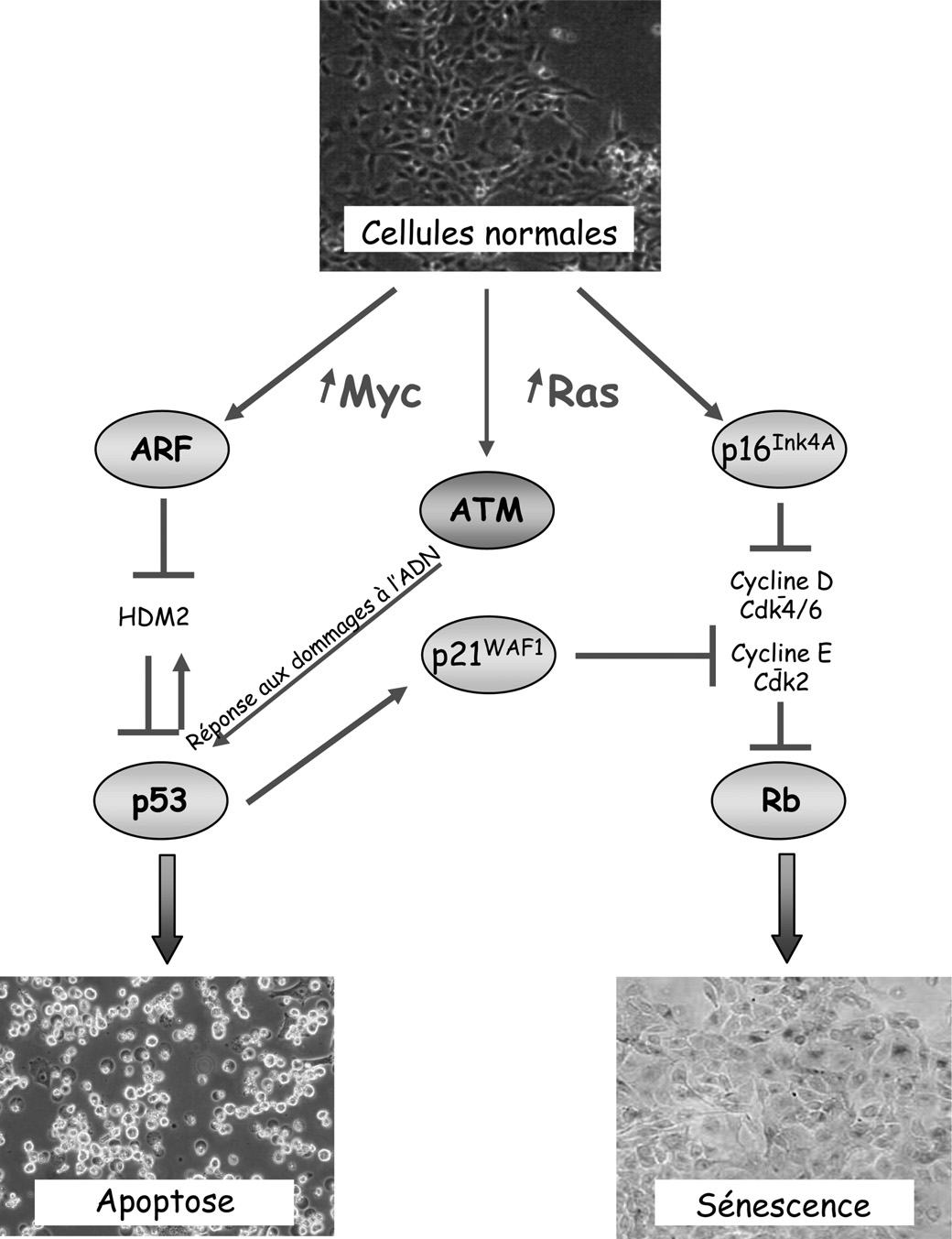
Fig. 2. — Représentation simplifiée des voies de signalisation impliquées dans l’induction de la sénescence et de la mort cellulaire par apoptose.
En réponse à l’activation d’oncogènes (Ras, Myc), l’induction des voies de signalisation dépendantes des protéines onco-suppressives p53 et Rb conduisent à la mort des cellules par apoptose ou à un arrêt définitif de la prolifération, un mécanisme connu sous le nom de sénescence prématurée. La protéine p53, également activée suite à des dommages subis par l’ADN ou suite à l’érosion naturelle des télomères (sénescence réplicative), est située à l’intersection entre les différentes voies de signalisation et joue donc un rôle prépondérant dans ces mécanismes de sauvegarde cellulaire.
La protéine p16INK4a est un inhibiteur des kinases dépendantes des cyclines (ou cdk) de la phase G qui inactivent Rb par phosphorylation. L’activation de Rb par p16INK4a conduit à une 1 sénescence cellulaire.
La protéine ARF ( Alternative Reading Frame ) interagit avec la protéine HDM2 et inhibe l’activité E3 nécessaire à l’ubiquitinylation de p53. En conséquence, p53 est stabilisée et peut accomplir ses fonctions d’arrêt du cycle cellulaire et d’induction de l’apoptose.
La protéine ATM est impliquée dans la reconnaissance des cassures double brin qui se produisent au cours de la prolifération anormale des cellules. Elle active la protéine p53 par phosphorylation.
La protéine p21WAF1 est un inhibiteur des kinases dépendantes des cyclines. Son expression est activée par p53, et elle se comporte comme un régulateur négatif du cycle cellulaire en phase G .
Avec l’objectif d’identifier des mécanismes d’inactivation fonctionnelle de p53, nous avons dans un premier temps étudié les neuroblastomes présentant une amplification de l’oncogène N-Myc [34]. Ces tumeurs, souvent métastatiques d’emblée, prolifèrent fortement en l’absence de mutations de p53 [35]. Nous avons ainsi mis en évidence une association étroite entre l’amplification de l’oncogène et la surexpression du gène Twist1 dans ces tumeurs, et démontré que Twist1 était capable d’inhiber la réponse apoptotique en présence de N-Myc, par inactivation de la voie dépendante de p53. Ainsi, en agissant comme facteur de survie, Twist1 coopère avec N-Myc pour la transformation maligne [17, 34]. Twist1 est capable d’inhiber la réponse de p53 à différentes étapes de sa régulation et de son activité: il bloque l’induction du suppresseur de tumeurs p14ARF, activateur de p53 en réponse aux oncoprotéines Myc ; il interagit physiquement et inactive HoxA5, un activateur transcriptionnel de p53 ; il inhibe certaines modifications post-traductionnelles (phosphorylations et acétylations) nécessaires à son activation [36, 37]. Enfin, Twist1 est capable d’interagir directement avec le domaine de liaison à l’ADN de p53 et d’altérer l’activation de ses gènes cibles [38]. En réponse à une activation aberrante des oncoprotéines Myc, Twist1 agit donc comme un véritable verrou de l’ensemble de la voie onco-suppressive médiée par p53.
L’expression de Twist1 et de son homologue structural et fonctionnel Twist2 est fréquemment réactivée dans les cancers humains, incluant un grand nombre de variétés de carcinomes (cancers du sein, du côlon, de l’œsophage, du pancréas, du foie, de la prostate…) [17, 18]. De façon intéressante, cette surexpression apparaît particulièrement fréquente dans les tumeurs présentant une activation de la voie Ras-MAPkinase. Comme mentionné précédemment, l’activation oncogénique de cette voie induit une réponse de type sénescence prématurée. Nous avons donc testé l’hypothèse selon laquelle les protéines Twist1 et Twist2 étaient capables de favoriser la transformation maligne en permettant aux cellules d’échapper à la sénescence. En accord avec cette hypothèse, nous avons montré que Twist1 et Twist2 coopèrent in vitro avec un mutant activé de Ras pour promouvoir la transformation maligne [18].
Les protéines Twist agissent en inhibant l’induction des inhibiteurs de cdk p21CIP1/WAF1etp16INK4A,deuxrégulateursclefsdesvoiesdépendantesdep53etdeRb.
La sénescence et l’apoptose sont des mécanismes onco-suppresseurs activés au cours des phases précoces du développement tumoral, en réponse à la détection d’une activité mitogénique anormale. L’inhibition de ces processus de sauvegarde est l’un des événements majeurs permettant l’émergence de cellules tumorales cancéreuses en réponse à un stress oncogénique. Longtemps restreinte à des observations réalisées dans des modèles de culture cellulaire, cette hypothèse repose désormais sur des données obtenues in vivo . Ainsi, les marqueurs de dommages à l’ADN et d’apoptose ont été mis en évidence dans les lésions précancéreuses de tumeurs de la vessie, du poumon, du sein et du colon. Ces marqueurs sont en revanche absents dans les lésions malignes correspondantes, suggérant fortement l’inhibition de la réponse apoptotique au cours de la progression vers la malignité [39, 40]. Par ailleurs, l’équipe de Manuel Serrano a pu montrer, dans un système expérimental murin de progression de tumeurs du poumon induites par l’oncogène K-RasV12, que l’évolution du stade d’adénome (bénin) au stade d’adénocarcinome (malin) s’accompagnait d’une perte des marqueurs de sénescence dans les lésions tumorales [41]. De même, l’inhibition de la sénescence prématurée est observée au cours de la progression maligne de différents types tumoraux induits par une activation aberrante de la voie Ras-MAPkinase, tels que les lymphomes ou les lésions tumorales mammaires malignes induits en réponse à l’oncogène H-RasV12 et les mélanomes induits par B-RafV600E [40, 42, 43]. Enfin, une sénescence prématurée peut également être observée en réponse à l’inhibition de certains mécanismes onco-suppresseurs, comme montré dans le modèle des tumeurs de la prostate induites par la perte du suppresseur de tumeur PTEN [44]. Ces travaux récents démontrent donc que les systèmes de sauvegarde cellulaire constituent effectivement in vivo une barrière capable de s’opposer à l’émergence de cellules à potentiel hyperprolifératif et que leur inhibition est nécessaire pour qu’une lésion pré- cancéreuse évolue en une lésion maligne. En accord avec un rôle dans l’échappement des cellules pré-cancéreuses à ces systèmes de sauvegarde, la surexpression des gènes Twist1 et/ou Twist2 est observée au cours de la transition entre les phases pré- maligne et maligne de la progression tumorale [18], appuyant un rôle crucial de ces facteurs au cours du développement de la tumeur primaire.
Échappement aux systèmes de sauvegarde cellulaire et EMT : deux processus intimement liés
En conditions expérimentales, la surexpression de Twist1 ou de Twist2 est suffisante pour abolir la sénescence normalement induite en réponse à une activation anormale des oncogènes Ras ou ErbB2. De façon intéressante, cet échappement cellulaire est associé à une TEM [18], avec acquisition de propriétés de motilité et d’invasion. Cette observation suggère que la réactivation de ces gènes embryonnaires favorise parallèlement la transition bénin-malin, par inhibition des systèmes de sauvegarde, et la dissémination métastatique, par induction d’une transdifférenciation (Figure 3). À l’appui de cette hypothèse, la dissémination des cellules pré- malignes dans un modèle tumoral mammaire murin a été observée dès le stade de l’hyperplasie, stade où l’expression de Twist1 est réactivée [45].
Très récemment, l’inhibition de p53 a été démontrée comme un événement favorisant la dédifférenciation cellulaire [46]. Par ailleurs, nous avons démontré, en parallèle avec le laboratoire de R. Weinberg, que le processus de TEM permettait aux cellules d’acquérir des propriétés de cellules souches, en particulier d’autorenouvellement [47, 48]. Ces observations suggèrent que les cellules souches cancé- reuses, cellules capables d’initier ou de régénérer une tumeur, peuvent non seulement provenir de la transformation maligne de cellules souches normales, mais aussi de la dédifférenciation de cellules différenciées par un processus de TEM. Du fait du rôle crucial joué par le microenvironnement dans le contrôle de ce processus, ces données suggèrent également que l’acquisition de cellules souches par les cellules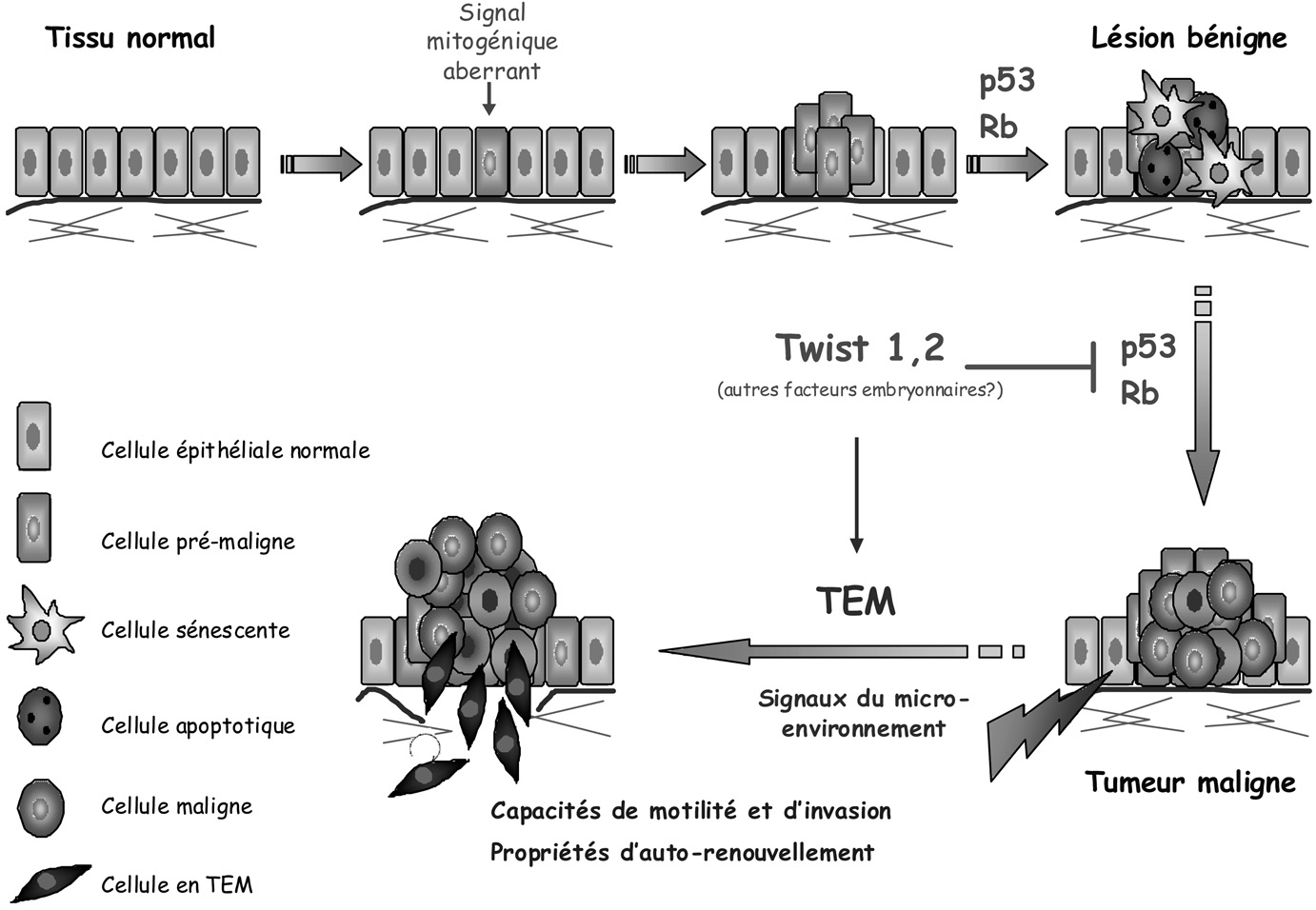
Fig. 3. — Inhibition précoce des mécanismes de sauvegarde et initiation du processus métastatique.
Dans les lésions pré-néoplasiques, l’activation d’oncogènes s’accompagne de l’induction des mécanismes de sauvegarde cellulaire que sont la sénescence et l’apoptose. L’expression des protéines Twist1 ou Twist2 permet d’inhiber ces mécanismes de sauvegarde et ainsi de favoriser la progression du stade bénin vers un stade malin. De plus, les protéines Twist provoquent une transition épithélio-mésenchymateuse (TEM), un processus associé à l’acquisition de capacités de motilité et d’invasion et de propriétés d’auto-renouvellement. L’inhibition des mécanismes de sauvegarde par Twist pourrait donc être directement associée à l’émergence de cellules cancéreuses présentant des caractéristiques de cellules métastatiques au cours des phases précoces de la progression tumorale.
pré-cancéreuses ou cancéreuses pourrait constituer un phénomène dynamique et instable.
CONCLUSION
L’existence d’un processus de TEM au cours de la progression tumorale a été longtemps contestée du fait de la difficulté technique à mettre en évidence des cellules ayant subi un processus de transdifférenciation au sein d’un échantillon tumoral. Aujourd’hui, de nombreuses observations, in vitro et in vivo , tendent à démontrer l’existence et l’implication de ce phénomène tant dans la croissance de la tumeur primaire que de la dissémination métastatique. Un nombre important de facteurs de transcription impliqués dans le contrôle de la TEM, dont Snail1, Snail2, Snail3, Zeb1, Zeb2, Foxc2 et Goosecoid, sont surexprimés de façon récurrente dans les cancers humains [19], suggérant que la propriété de Twist1 et Twist2 de promou- voir la transition entre les stades bénin et malin et de favoriser la dissémination précoce des cellules cancéreuses est partagée par d’autres facteurs de transcription d’origine embryonnaire. Cette hypothèse implique une intrication fonctionnelle entre les mécanismes d’échappement aux systèmes de surveillance cellulaires et la TEM. L’inactivation de Rb et de p53 favorise l’induction d’une TEM en présence d’une activité mitogénique de nature oncogénique et augmente de façon significative l’efficacité de la dédifférenciation, en conditions expérimentales, de cellules somatiques en cellules pluripotentes. Ce dernier effet est probablement lié à la protection apportée aux cellules contre les processus de sénescence et d’apoptose [46]. Aussi, l’échappement aux systèmes de sauvegarde cellulaire et la TEM apparaissent comme deux processus présentant une forte dépendance réciproque. L’inhibition des voies mettant en jeu les protéines p53 et par Rb favorise la TEM, tandis que certains des facteurs de transcription impliqués dans le contrôle de la TEM contribuent à une perte des fonctions onco-suppressives. La résultante de ces processus est la génération de cellules présentant une importante plasticité, caractérisée par une perte des contraintes onco-suppressives, l’acquisition de propriétés d’autorenouvellement, de motilité et d’invasion et la possibilité de s’adapter à un nouveau microenvironnement. Enfin, comme la dédifférenciation cellulaire confère aux cellules des propriétés de résistance accrue aux drogues cytotoxiques et génotoxiques, la TEM pourrait également favoriser les processus de chimiorésistance et de récidive. En accord avec cette hypothèse, les traitements chimiothérapeutiques conduisent, in vivo , à une sélection de cellules tumorales circulantes exprimant des facteurs de transcription embryonnaires et des marqueurs mésenchymateux [49]. Aussi, il apparaît légitime de conceptualiser des molécules capables de cibler efficacement ces cellules [50]. De nouvelles approches basées sur l’utilisation d’oligonucléotides modifiés, capables d’empêcher la liaison à l’ADN des facteurs de transcription Snail1, Snail2 et Zeb2 ont été conçus récemment à cet effet. Ces expériences pionnières pourraient ouvrir la voie à des innovations thérapeutiques prometteuses dont l’objectif serait de contrecarrer tout à la fois le développement de la tumeur primaire et le processus de dissémination métastatique.
BIBLIOGRAPHIE [1] Hanahan D., Weinberg R.A. — The hallmarks of cancer.
Cell., 2000, 100 , 57-70.
[2] Folkman J. — Tumor angiogenesis: therapeutic implications.
N. Engl. J. Med., 1971, 285 , 1182- 1186.
[3] Geiger T.R., Peeper D.S. — Metastasis mechanisms. Biochim. Biophys. Acta., 2009, 1796 , 293-308.
[4] Fukumura D., Xavier R., Sugiura T. et al. — Tumor induction of VEGF promoter activity in stromal cells.
Cell, 1998, 94 , 715-725.
[5] Cahill D.P., Kinzler K.W., Vogelstein B. et al. — Genetic instability and darwinian selection in tumours.
Trends Cell Biol. , 1999, 9 , M57-M60.
[6] McGowan P.M., Kirstein J.M., Chambers A.F. — Micrometastatic disease and metastatic outgrowth: clinical issues and experimental approaches. Future Oncol., 2009, 5 , 1083-1098.
[7] Thiery J.P., Sleeman J.P. — Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell. Biol. , 2006, 7, 131-142.
[8] Nakaya Y., Sheng G. — Epithelial to mesenchymal transition during gastrulation: an embryological view. Dev. Growth Differ., 2008, 50 , 755-766.
[9] Iwano M., Plieth D., Danoff T.M. et al . — Evidence that fibroblasts derive from epithelium during tissue fibrosis.
J. Clin. Invest., 2002, 110 , 341-350.
[10] Berx G., Raspe E., Christofori G. — Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin. Exp. Metastasis, 2007, 24 , 587-597.
[11] Zavadil J., Bottinger E.P. — TGF-beta and epithelial-to-mesenchymal transitions.
Onco- gene, 2005, 24 , 5764-5774.
[12] Peinado H., Quintanilla M., Cano A. — Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J.
Biol. Chem., 2003, 278 , 21113-21123.
[13] Logan C.Y., Nusse R. — The Wnt signaling pathway in development and disease.
Annu. Rev.
Cell. Dev. Biol., 2004, 20 , 781-810.
[14] Peinado H., Olmeda D., Cano A. — Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer, 2007, 7 , 415-428.
[15] Cannito S, Novo E, Compagnone A, et al. — Redox mechanisms switch on hypoxiadependent epithelial-mesenchymal transition in cancer cells.
Carcinogenesis, 2008, 29 , 2267- 2278.
[16] Yang M.H., Wu M.Z., Chiou S.H., et al. — Direct regulation of TWIST by HIF-1alpha promotes metastasis.
Nat. Cell. Biol. , 2008, 10 , 295-305.
[17] Puisieux A., Valsesia-Wittmann S., Ansieau S. — A twist for survival and cancer progression.
Br. J. Cancer, 2006, 94 , 13-17.
[18] Ansieau S., Bastid J., Doreau A. et al. — Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence.
Cancer Cell, 2008 ; 14 , 79-89.
[19] Yang J., Weinberg R.A. — Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell, 2008, 14 , 818-829.
[20] Yang J., Mani S.A., Donaher J.L. et al. — Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis.
Cell, 2004, 117 , 927-939.
[21] Matsuo N., Shiraha H., Fujikawa T. et al. — Twist expression promotes migration and invasion in hepatocellular carcinoma.
BMC Cancer, 2009, 9 , 240.
[22] Oka H., Shiozaki H., Kobayashi K. et al. — Expression of E-cadherin cell adhesion molecules in human breast cancer tissues and its relationship to metastasis.
Cancer Res., 1993, 53 , 1696-1701.
[23] Hirata T., Fukuse T., Naiki H. et al. — Expression of E-cadherin and lymph node metastasis in resected non-small-cell lung cancer.
Clin. Lung Cancer, 2001, 3 , 134-140.
[24] Kaihara T., Kusaka T., Nishi M. et al. — Dedifferentiation and decreased expression of adhesion molecules, E-cadherin and ZO-1, in colorectal cancer are closely related to liver metastasis. J. Exp. Clin. Cancer Res., 2003, 22 , 117-123.
[25] Onder T.T., Gupta P.B., Mani S.A. et al. — Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways.
Cancer Res. 2008, 68 , 3645-3654.
[26] Tarin D., Thompson E.W., Newgreen D.F. — The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res., 2005, 65 , 5996-6000.
[27] Spaderna S., Schmalhofer O., Hlubek F. et al. — A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer.
Gastroenterology, 2006, 131 , 830-840.
[28] Trimboli A.J., Fukino K., De Ba. et al. — Direct evidence for epithelial-mesenchymal transitions in breast cancer.
Cancer Res., 2008, 68 , 937-945.
[29] Lowe S.W., Cepero E., Evan G. — Intrinsic tumour suppression.
Nature, 2004, 432 , 307-315.
[30] Vousden K.H., Lane D.P. — p53 in health and disease.
Nat. Rev. Mol. Cell. Biol, 2007, 8 , 275-283.
[31] Evan G.I., Wyllie A.H., Gilbert C.S. et al. — Induction of apoptosis in fibroblasts by c-myc protein.
Cell, 1992, 69 , 119-128.
[32] Eischen C.M., Weber J.D., Roussel M.F. — Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. , 1999, 13 , 2658-2669.
[33] Serrano M., Lin A.W., McCurrach M.E. et al. — Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a.
Cell, 1997, 88 , 593-602.
[34] Valsesia-Wittmann S., Magdeleine M., Dupasquier S. et al. — Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells.
Cancer Cell, 2004, 6 , 625-630.
[35] Brodeur G.M., Seeger R.C., Schwab M. et al. — Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage.
Science, 1984, 224 , 1121-1124.
[36] Maestro R., Dei Tos A.P., Hamamori Y. et al. — Twist is a potential oncogene that inhibits apoptosis.
Genes Dev. ,1999, 13 , 2207-2217.
[37] Stasinopoulos I.A., Mironchik Y., Raman A. et al . — HOXA5-twist interaction alters p53 homeostasis in breast cancer cells.
J. Biol. Chem., 2005, 280 , 2294-2299.
[38] Shiota M., Izumi H., Onitsuka T. et al. — Twist and p53 reciprocally regulate target genes via direct interaction.
Oncogene, 2008.
[39] Gorgoulis V.G., Vassiliou L.V., Karakaidos P. et al. — Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions.
Nature, 2005, 434 , 907-913.
[40] Bartkova J., Rezaei N., Liontos M. et al. — Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints.
Nature, 2006, 444 , 633-637.
[41] Collado M., Gil J., Efeyan A. et al. — Tumour biology: senescence in premalignant tumours.
Nature, 2005, 436 , 642.
[42] Sarkisian C.J., Keister B.A., Stairs D.B. et al . — A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell. Biol. 2007, 9 , 493-505.
[43] Michaloglou C., Vredeveld L.C., Soengas M.S. et al. — BRAFE600-associated senescencelike cell cycle arrest of human naevi.
Nature, 2005, 436 , 720-724.
[44] Chen Z., Trotman L.C., Shaffer D. et al. — Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis.
Nature, 2005, 436 , 725-730.
[45] Husemann Y., Geigl J.B., Schubert F. et al. — Systemic spread is an early step in breast cancer.
Cancer Cell, 2008, 13, 58-68.
[46] Marion R.M., Strati K., Li H. et al. — A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity.
Nature, 2009, 460 , 1149-1153.
[47] Morel A.P., Lievre M., Thomas C. et al. — Generation of breast cancer stem cells through epithelial-mesenchymal transition.
PLoS ONE, 2008, 3, e2888.
[48] Mani S.A., Guo W., Liao M.J. et al. — The epithelial-mesenchymal transition generates cells with properties of stem cells.
Cell , 2008, 133, 704-715.
[49] Creighton C.J., Li X., Landis M. et al. — Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features.
Proc. Natl. Acad. Sci. U S A. 2009, 106 , 13820-13825.
[50] Gupta P.B., Onder T.T., Jiang G. et al. — Identification of selective inhibitors of cancer stem cells by high-throughput screening.
Cell ., 2009, 138 , 645-659.
DISCUSSION
M. Bernard PESSAC
Quel est le niveau de synthèse et d’excrétion de glycosaminoglycanes dans les cellules CD44 high ?
À ma connaissance, ce point n’a pas été étudié de façon spécifique. Cependant, certaines équipes ont montré que l’hyaluronane (HA), glycosaminoglycane majeure de la matrice extra-cellulaire, était exprimée dans les cellules épithéliales cancéreuses, son expression étant corrélée au potentiel métastatique des tumeurs. HA est capable de se lier à CD44, ce qui est à l’origine de l’activation de différentes voies de signalisation telles que celles activées par le TGF-β ou ErbB2. CD44 étant exprimé dans les cellules tumorales mammaires susceptibles de subir une TEM avec acquisition de propriétés de cellules souches, l’étude du rôle de l’interaction HA-CD44 apparaît tout à fait pertinente dans ce modèle.
M. Maurice TUBIANA
Au terme de cette passionnante séance, j’avoue être un peu perplexe. D’une part, certaines données suggèrent que la TEM, qui est un phénomène fondamental pendant l’embryogenèse, est aussi un phénomène fréquent en dehors d’elle, à la fois pendant les processus physiologiques (réparation d’une plaie), pathologiques (fibrose, cancer) et pendant les études in vitro et qu’il peut être provoqué par un grand nombre de facteurs (hypoxie, inflammation, etc.). Inversement, d’autres arguments suggèrent que la TEM est réversible dans les phénomènes physiologiques et non ou peu réversible dans les processus pathologiques et que les signaux qui provoquent ces TEM irréversibles ou peu réversibles (par exemple dans la cancérogenèse) sont complexes, peu fréquents et relativement spécifiques. Merci d’essayer de clarifier ce problème ?
Comme indiqué par Jean-Paul Thiery, le rôle de la TEM au cours de l’embryogenèse est parfaitement décrit. En revanche, dans les processus pathologiques tels que les cancers, son existence et son rôle ont fait l’objet de vives controverses car il est difficile de différencier les cellules épithéliales ayant subi une TEM, des cellules stromales environnantes. On dispose cependant aujourd’hui d’arguments in vitro , mais aussi, in vivo , en faveur de l’existence de ce processus de transdifférenciation au cours de différentes phases de la progression tumorale. Malgré tout, comme vous le soulignez à juste titre, in vivo les facteurs déclenchant restent à déterminer.
In vitro , l’activation de voies de signalisation telles que celles activées par les cytokines et certains récepteurs de type tyrosine kinase, mais aussi l’hypoxie ou des contraintes physiques peuvent induire une TEM. Ces différentes voies sont toutes impliquées dans la cancérogenèse humaine mais il reste impossible à ce jour d’identifier laquelle de ces voies (ou la conjonction de quelles voies) est impliquée dans une tumeur donnée. Il est tout à fait clair qu’en dépit de la génération de nouveaux modèles animaux, la plupart des travaux effectués restent limités à des études in vitro , ce qui limite considérablement nos connaissances concernant les mécanismes mis en jeu au cours de la progression tumorale chez l’homme. Concernant la réversibilité de la TEM, elle peut avoir lieu tant au niveau du développement embryonnaire qu’au cours de la progression tumorale. Ainsi, si on estime que la TEM facilitera l’acquisition de capacités de motilité cellulaire, d’invasion et d’intravasation, différents articles récents montrent qu’un processus de TME (transition mésenchymo-épithéliale) est nécessaire à la formation de macrométastases dans le tissu secondaire. Ceci est cohérent avec l’observation selon laquelle une métastase présente généralement un phénotype similaire (épithélial) à celui de la tumeur primaire. En conclusion, la TEM est un processus hautement dynamique car dépendant du micro-environnement, et donc réversible que ce soit dans un contexte physiologique ou pathologique.
M. Jacques ROUE ı
SSE
La transition épithélio-mésenchymateuse est-elle un évènement propre aux tumeurs malignes ou existe-t-elle dans les tumeurs bénignes ou dans les cancers in situ ?
Il s’agit d’une question très importante. La TEM est présentée comme un processus favorisant la dissémination métastatique, et on l’envisage de ce fait généralement comme un processus assez tardif. Cependant, comme je l’ai montré au cours de mon exposé, nos travaux montrent qu’en permettant aux cellules d’échapper à la sénescence et à l’apoptose et d’acquérir certaines propriétés de cellules souches, elle pourrait aussi faciliter la croissance de la tumeur primaire. À ce titre, les travaux de l’équipe de Christoph Klein ont démontré, sur la base de modèles de souris transgéniques de cancers mammaires, que certaines cellules pouvaient envahir les tissus adjacents et entrer dans la circulation dès le stade de l’hyperplasie atypique. De façon intéressante, la protéine Twist1, facteur de transcription activant le processus de TEM, est surexprimé dans ce modèle dès ce stade de la progression tumorale. Il est donc tout à fait plausible que des processus de TEM puissent survenir dans certaines lésions pré-malignes.
M. Claude DREUX
Est-il possible d’utiliser ces facteurs de transcription impliqués dans l’induction de la TEM comme des cibles thérapeutiques ?
Au plan théorique, ces facteurs constituent en effet d’excellentes cibles thérapeutiques car, d’une part, ils sont surexprimés dans les cellules cancéreuses mais non exprimés dans la grande majorité des cellules adultes normales, et, d’autre part, leur inactivation devrait permettre tout à la fois de freiner la croissance de la tumeur primaire et de prévenir la dissémination métastatique. Cependant, au-delà même des problèmes de vectorisation, ces approches sont rendues difficiles par deux phénomènes. Ce sont des protéines nucléaires, qui ne sont pas les protéines les plus faciles à cibler. De plus, dans une tumeur donnée, il apparaît que plusieurs de ces facteurs sont généralement réactivés pour orchestrer le processus de TEM. Il est donc vraisemblable que l’inactivation ciblée d’un facteur donné (bien que produisant une sénescence ou une apoptose dans des systèmes in vitro ) présente un effet limité in vivo . Seules des approches permettant leur inhibition conjointe pourraient donc être envisagées. De manière intéressante, notons que dans des modèles cellulaires des versions modifiées d’oligonucléotides sont capables d’inhiber de manière conjointe la fixation des protéines SNAI1, SNAI2 et ZEB2 sur leurs séquences cibles, offrant ainsi une première approche de ce type. Une approche distincte consisterait à cibler spécifiquement les cellules ayant subi une TEM sur la base d’un différentiel d’expression d’antigènes ou de sensibilité à certaines drogues.
* Centre Léon Bérard, Lyon, F-69008 ; Inserm, U590, Lyon, F-69008 ; Université de Lyon, Lyon 1, ISPB, Lyon, F-69003, France, e-mail : puisieux@lyon.fnclcc.fr. Tirés-à-part : Professeur Alain Puisieux, même adresse. Article reçu et accepté le 23 novembre 2009.
Bull. Acad. Natle Méd., 2009, 193, no 9, 2017-2034, séance du 1er décembre 2009