
Résumé
La pharmacogénétique étudie les effets des variations individuelles de la séquence de l’ADN sur la réponse aux médicaments. L’identification des facteurs génétiques qui contrôlent l’activité des enzymes du métabolisme des médicaments, les cytochromes P450 (CYP) a constitué son premier champ d’application chez l’homme. Trois familles de ces enzymes sont responsables du métabolisme des médicaments. Leurs gênes ont été identifiés et il existe pour certains d’entre eux un polymorphisme génétique dans la population permettant de séparer les sujets en métaboliseurs rapides homozygotes sauvages (porteurs de deux allèles sauvages) et métaboliseurs lents (allèles mutés) hétérozygotes ou homozygotes mutés. Le CYP2C9 est l’enzyme qui métabolise les anticoagulants coumariniques comme l’acenocoumarol ou la warfarine. Un génotype homozygote muté CYP2C9 *3/ *3 est présent dans 0,7 % de la population caucasienne et correspond à un phénotype métaboliseur lent entraî- nant une accumulation de ces médicaments dans l’organisme, un effet anticoagulant important et un risque hémorragique. Au cours de trois études cliniques nous avons pu montrer que la présence d’au moins un variant allèlique CYP2C9 *3 chez des patients ou des sujets volontaires sains expliquait 14 % de la variabilité individuelle de la réponse au traitement. Une quatrième étude portant sur le génotype du site récepteur des anticoagulants, la vitamine K époxyde réductase, a montré que l’association génotypique du variant allèlique CYP2C9 *3 et d’un SNP du site récepteur vitamine K epoxyde réductase (VKORC1) expliquait 50 % de la variabilité individuelle de la réponse au traitement anticoagulant. L’association de ces deux mutations chez un individu risque donc d’entraîner une importante réponse au traitement anticoagulant avec un risque hémorragique élevé. Cet exemple montre la voie de développement de la pharmacogénétique chez l’homme lorsqu’après avoir identifié des médicaments à haut risque, on découvre leurs voies métaboliques préférentielles et l’existence de mutations dans les gènes qui contrôlent leur expression. L’étude du rôle des facteurs génétiques dans la variabilité de la réponse aux traitements ne fait que commencer .
Summary
Pharmacogenetics, a discipline still in its infancy, is the study of genetically determined variations in how individuals respond to drugs. Mutations may affect drug metabolism, transmembrane transport into cells, or target receptors. Genetic polymorphisms affecting drug metabolism were the first to be identified. Genetic factors control the activity of phase I reactions involving cytochrome (CYP) P450 isoenzymes. Three CYP families catalyze drug metabolism in humans. Their genes have been identified and polymorphisms have been described in various populations, leading to either high activity (‘‘ extensive metabolizer ’’ phenotype) or low activity (‘‘ poor metabolizer ’’ phenotype).The CYP2C9 polymorphism illustrates the potential clinical importance of pharmacogenetics. This enzyme catalyzes the metabolism of the coumarinic oral anticoagulants acenocoumarol and warfarin. The homozygous mutant genotype CYP 2C9 * 3/*3 , present in 0,7 % of Caucasians, leads to low enzyme activity and thus to the accumulation of these drugs in the body ; this in turn increases the anticoagulant activity and induces a higher risk of bleeding. In three clinical studies of patients and healthy volonteers, we found that this CYP2C9 *3 mutant allele was responsible for 14 % of the variability in the response to these drugs. Then, by studying the genetic polymorphism of the receptor site of oral anticoagulants — the vitamin K epoxide reductase multiprotein complex — we showed that a combination of the two genetic variants (CYP2C9 and the receptor site) was responsible for 50 % of the variability. These data suggest that patients who have both genetic polymorphisms could be at an increased risk of bleeding during oral anticoagulant therapy.
La Pharmacogénétique est l’étude des effets des variations individuelles de la séquence de l’ADN sur la réponse aux médicaments. On la distingue aujourd’hui de la Pharmacogénomique qui, d’un point de vue plus vaste, étudie l’influence de la variabilité individuelle de l’expression des gènes impliqués dans la susceptibilité aux maladies et la réponse aux médicaments au niveau d’une cellule, d’un tissu, d’un individu ou d’une population.
C’est au milieu du siècle dernier qu’émerge le concept d’une variabilité d’origine génétique dans la réponse aux médicaments. Le premier type de variabilité observé concerne le contrôle génétique de l’activité des enzymes du métabolisme des médicaments. En 1953 le phénotype acétyleur lent de l’isoniazide est pour la première fois décrit par Bonicke et Reif [1] en Allemagne et Hugues et coll. [2] aux
USA. Ces derniers, l’année suivante, montrent que les sujets acétyleurs lents ont
plus de risque de présenter une neuropathie périphérique à l’isoniazide que les sujets acétyleurs rapides [3]. En 1956 Carson et coll . [4] découvrent qu’un déficit en glucose-6-phosphate déshydrogènase est à l’origine des crises hémolytiques déclenchées par un antipaludéen, la primaquine, et en 1957 Kalon et Genest [5] montrent que les accidents respiratoires de la succinylcholine surviennent chez des patients déficitaires en sérum cholinestérase. Le concept d’un phénotype ‘‘ métaboliseur lent ’’ était né. On sait à l’époque que ces phénotypes sont liés à des facteurs génétiques et se transmettent sur un mode mendelien. On peut identifier certains patients qui présentent des effets indésirables liés à des concentrations plasmatiques très élevées des médicaments et à l’absence de métabolites dans les urines. Mais les techniques de biologie moléculaire ne permettent pas encore de localiser les gènes responsables.
Il faudra attendre, dans les années 1960-70, l’identification progressive des principaux enzymes du métabolisme, les cytochromes P450 (CYP) pour caractériser les voies métaboliques défaillantes chez ces patients métaboliseurs lents. Puis on identifie les gènes codant pour les différentes protéines qui sont préalablement identifiées et purifiées. Les polymorphismes génétiques responsables des phénotypes métaboliseurs lents sont alors un à un découverts. Finalement c’est le séquençage du génome humain qui va permettre de développer de façon spectaculaire l’analyse des variants allèliques impliquant soit un seul nucléotide (SNP ou single nucleotide polymorphism) soit le nombre de répétitions en tandem dans une séquence plus ou moins longue (micro ou mini-satellite).
Le développement considérable de la pharmacogénétique, attesté par la progression exponentielle des publications qui y sont consacrées, couvre aujourd’hui trois grands domaines, tous impliqués dans la variabilité interindividuelle de la réponse aux traitements : les enzymes du métabolisme des médicaments, les transporteurs transmembranaires des médicaments et les récepteurs ou sites effecteurs des médicaments. En effet les variations d’origine génétique de l’activité des transporteurs transmembranaires et des récepteurs des médicaments sont l’objet d’une recherche plus récente mais tout aussi importante.
L’objectif de cette présentation est, en fondant le discours sur les travaux de notre équipe sur le CYP2C9 , de montrer comment les variations génétiques de l’expression de cette protéine ainsi que celles du site récepteur d’un médicament anticoagulant, l’acenocoumarol (SINTROM®) peuvent modifier la réponse aux anticoagulants coumariniques, faisant courir à certains patients un risque élevé d’hémorragies ou les exposant au risque d’interactions médicamenteuses. Notre conclusion portera d’une manière plus générale sur l’intérêt du développement des études pharmacogénétiques et sur l’importance de la prise en compte de ces résultats par le praticien lors de la prescription de certains médicaments.
LES ENZYMES DU MÉTABOLISME DES MÉDICAMENTS
Les médicaments sont des corps étrangers qui sont introduits dans l’organisme humain par différentes voies (résorption intestinale ou transcutanée ou transmuqueuse), vont circuler dans le sang et les tissus, agir sur les récepteurs où ils déclenchent des effets thérapeutiques puis être éliminés de l’organisme. Certains le sont directement dans les urines sans subir de biotransformation. La plupart sont en fait métabolisés, principalement dans le foie. Ce processus de biotransformation aboutira à des métabolites généralement hydrosolubles qui seront éliminés soit dans la bile et les matières fécales, soit dans les urines après avoir recirculé dans le sang du foie jusqu’aux reins.
Le métabolisme de ces médicaments correspond schématiquement à deux étapes :
les enzymes de phase I ou enzymes de fonctionnalisation, les CYP, réalisent la première étape du métabolisme des médicaments dont les métabolites peuvent soit être éliminés directement s’ils sont solubles dans les milieux aqueux (bile, urine), soit nécessiter une seconde étape prise en charge par les enzymes de phase II, les transférases. Ces transférases en opérant une conjugaison (methylation, acétylation, glycuro ou glutathion conjugaison) vont aboutir à des métabolites hydrosolubles plus facilement éliminables.
Les CYP sont des hémoprotéines identifiées selon une nomenclature proposée en 1987 par NEBERT et collaborateurs et toujours utilisée aujourd’hui [6]. En fonction du pourcentage de séquence d’acides aminés identiques d’une protéine à l’autre, on distingue des familles (≥ 40 % d’identité) marquées par un chiffre arabe (exemple :
les familles des CYP 1, 2, 3 qui métabolisent tous les médicaments), puis des sous-familles (≥ 55 % d’identité) qui sont désignées par une lettre (
CYP 1A, 2D, 3A etc). Enfin un nombre arabe identifie une protéine donnée isoforme, par exemple le CYP 3A4 ou le CYP2C9. Sur les 270 familles génétiques de CYP connues à ce jour, 18 sont exprimées chez les humains avec 42 sous-familles [6]. Les familles des
CYP1, 2 et 3 métabolisent les médicaments, alors que le CYP4 est impliqué dans le métabolisme de divers types d’acides gras polyinsaturés tels que l’acide arachidonique, le CYP5 est la thromboxane A2 synthase, le CYP 8 est la prostacycline synthase, etc…
Ces protéines sont synthétisées à partir d’un code génétique contenu dans les exons de certains gênes. On sait aujourd’hui que les individus humains avec leurs 30 000 gènes varient les uns les autres de trois à huit millions de paires de bases sur un total de l’ordre de trois milliards. Tous nos gènes sont polymorphes et la plupart de ces variations de séquences d’un individu à l’autre consistent en un remplacement d’une base par une autre (SNP) dans une position donnée. Plus rarement on observe des délétions d’une base ou des insertions d’une ou plusieurs bases. On ne parle de mutation que pour caractériser des modifications génétiques très rares dans une population (moins de 1 % des sujets). En revanche, on dit qu’il existe un polymorphisme génétique lorsque la fréquence de ces modifications génétiques est supé-
rieure ou égale à 1 % dans une population donnée. Seule une petite minorité de ces modifications génétiques auront une traduction clinique lorsqu’un ou plusieurs SNP altèrent le niveau d’expression de la protéine ou son activité. Une anomalie du génotype d’un CYP se traduira par un phénotype particulier de métabolisation du médicament s’il n’existe pas d’autre voie métabolique de remplacement dans l’organisme et ce phénotype n’aura d’importance pour les cliniciens que s’il entraîne une variation importante de la réponse de l’organisme au médicament (perte d’efficacité ou toxicité).
Cette relation entre un génotype et un phénotype particulier doit être systématiquement explorée pour chaque médicament chez l’homme. La recherche de SNP dans le génome repose sur des techniques d’identification des bases mutées permettant de définir les deux allèles. L’étude du phénotype de métabolisation repose sur l’exploration de la biotransformation de médicaments tests (probe drugs) dont on connaît la ou les voies métaboliques préférentielles chez l’homme et qui permettent d’analyser dans le sang ou les urines, le degré de métabolisation (rapport des concentrations du médicament parent sur celles du métabolite principal). Ceci permet d’identifier un polymorphisme génétique dans une population, c’est-à-dire de classer les individus en métaboliseurs lents (déficit métabolique) lié à un ou plusieurs SNP et en métaboliseurs rapides, voire même en métaboliseurs ultrarapides pour le CYP 2D6.
Lorsque le génotype et le phénotype de métabolisation concordent bien, on estime que le génotype (plus facile à réaliser aujourd’hui) a une forte valeur prédictive positive.
LE CYTOCHROME P450 2C9
Le cytochrome P450 2C9 (
CYP2C9 ) est principalement exprimé dans les hépatocytes où il contrôle le métabolisme des anticoagulants coumariniques, d’un antiépileptique la phenytoine, d’un antidiabétique le tolbutamide, de certains antiinflammatoires non stéroidiens et d’un antagoniste des récepteurs de l’angiotensine de type I, le losartan. Des travaux récents de notre équipe indiquent que le CYP2C9 métabolise également les autres anticoagulants oraux, dérivés de l’indane-dione, comme le fluindione (Previscan®), très utilisé en France. En terme quantitatif, ce CYP représente 20 % de l’ensemble des CYP hépatiques responsables du métabolisme des médicaments [7]. Deux variants allèliques du
CYP2C9 ont été observés (*2 et *3) à coté de la forme dite ‘‘ sauvage ’’ ( * 1) dans la population. Ces variants *2 et *3 correspondent à un phénotype métaboliseur lent pour les médicaments métabolisés par le CYP2C9 . Le variant CYP2C9 *2 correspond au remplacement d’une arginine par une cystéine en position 144 de la protéine du
CYP2C9 et le variant *3 correspond au remplacement d’une isoleucine par une leucine en position 359. La fréquence des allèles sauvages et mutés est présentée dans le tableau 1 où nous avons comparé plusieurs populations [8, 9]. Dans la population française, le génotype sauvage homozygote *1/*1 est présent chez 62 % des individus, et la fréquence des génotypes homozygotes mutés *2/*2 et *3/*3 est respectivement de 2,7 % et 0,7 %.
TABLEAU 1. — Fréquence des allèles du CYP 2C9 dans trois populations (A : France n = 151, B :
Chine n = 394, C : Danemark n = 276) [ref. 8,9] Allèle Fréquence A B C CYP 2C9 *1 0,77 (0,72-0,82) 0,963 (0,945-0,975) 0,826 (0,792-0,857) CYP 2C9 *2 0,15 (0,11-0,19) 0,001 (0-0,007) 0,121 (0,095-0,152) CYP 2C9 *3 0,08 (0,05-0,12) 0,036 (0,04-0,051) 0,053 (0,035-0,075) CYP 2C9 ET METABOLISME DES ANTICOAGULANTS COUMARINIQUES
Les anticoagulants coumariniques, warfarine (Coumadine®) et acenocoumarol (Sintrom®) sont métabolisés par le CYP2C9 [10]. On sait que les doses de warfarine [11] et d’acenocoumarol [12] nécessaires pour maintenir l’INR dans les limites d’une anticoagulation efficace mais suffisante sont plus faibles chez les patients homozygotes mutés CYP2C9 *3/*3. Trois cas de surdosage en warfarine ont été décrits dans les premiers jours du traitement entraînant une diminution considérable de la dose de warfarine nécessaire pour obtenir une anticoagulation efficace [11, 13].
Nous avons été les premiers à étudier, à l’hôpital St-Antoine le risque de surdosage à l’acenocoumarol. Cette étude a permis à C.VERSTUYFT et coll. [14] d’observer deux cas cliniques typiques liés à la présence du variant allèlique homozygote CYP2C9 * 3/ *3. Le premier cas concernait une jeune fille âgée de 18 ans, hospitalisée pour une intervention orthopédique sur le genou et traitée préventivement par une dose standard (un comprimé à 4 mg par jour) d’acenocoumarol. Au troisième jour du traitement, l’INR était égal à 9 avec épistaxis et gingivorragie. Après une interruption transitoire du traitement, il suffira d’une dose huit fois plus faible d’acenocoumarol pour maintenir l’INR entre 2 et 3. Le deuxième cas concernait une femme âgée de 82 ans traitée par l’héparine avec relais par l’acenocoumarol pour une arythmie complète par fibrillation auriculaire. Au quatrième jour de traitement un INR supérieur à 9 sans signe hémorragique a nécessité une diminution drastique des doses d’acenocoumarol (0,5 mg/jour) pour stabiliser l’INR entre 2 et 3. Aucun autre facteur concomitant hépatique ou thyroïdien n’expliquait ce surdosage dans les deux cas et il n’y avait pas d’interaction médicamenteuse en cause. Ce sont les deux premiers cas de surdosage à l’acenocoumarol observés chez des patients homozygotes mutés CYP2C9 *3/*3.
À la suite de ces observations, nous avons entrepris une étude prospective cascontrôle [15] au cours de laquelle ont été recherchés les différents facteurs de risque d’un surdosage en anticoagulant. Soixante-quinze patients consécutifs ayant un INR > 4 ont été comparés à 75 sujets témoins appariés sur l’âge, l’anticoagulant oral
et la dose quotidienne d’anticoagulant, ayant un INR compris entre 2 et 3,5. Les principaux facteurs génétiques et environnementaux impliqués dans un éventuel surdosage coumarinique ont été passés en revue. Les résultats de ce travail montrent qu’une interaction médicamenteuse et une récente augmentation des doses d’anticoagulant sont les deux principaux facteurs environnementaux expliquant le surdosage avec une augmentation du risque de 2,13 (IC95 % : 1,06 — 7,57) et de 3,38 (IC95 % : 1,51 — 7,57) respectivement. Trois patients homozygotes mutés CYP2C9 *3/*3 ont été observés dans le groupe des INR > 4 mais aucun dans le groupe des
INR compris entre 2 et 3,5. Malheureusement les effectifs de cette étude étaient insuffisants pour mettre en évidence, statistiquement parlant, l’impact réel du polymorphisme génétique du CYP2C9 sur le risque de surdosage.
Ces résultats nous ont amené à réaliser un troisième travail [16] chez 263 sujets volontaires sains. Ils ont tous reçu une dose unique de 4 mg d’acenocoumarol et nous avons analysé leur génotype du CYP2C9 en corrélant les résultats du génotypage à ceux d’un phénotype correspondant à l’effet anticoagulant (baisse de l’activité du facteur VII, proconvertine, dans le plasma vingt-quatre heures après la prise du médicament).Les résultats de cette étude montrent que la présence d’un allèle muté CYP2C9 *3 était le seul facteur génétique expliquant la variation de la réponse pharmacodynamique sur le facteur VII dont l’activité diminuait à 60 fi 19 % chez les homozygotes sauvages CYP2C9 *1/*1, à 39 fi 17 % chez les hétérozygotes
CYP2C9 *1/*3, et à 17 % pour les homozygotes mutés CYP2C9 *3/*3 (p = 0,001). La présence d’un ou deux allèles mutés du
CYP2C9 était donc responsable d’un phénotype métaboliseur lent, expliquant l’effet anticoagulant plus important de l’acenocoumarol. La présence de l’allèle muté CYP2C9 *3 expliquait 14 % de la variabilité interindividuelle de la réponse au traitement anticoagulant.
Ces observations confirment que la présence d’un ou deux allèles mutés CYP2C9 *3 entraîne un risque de plus forte activité anticoagulante de l’acenocoumarol et donc un risque hémorragique plus élevé si les patients sont traités par des doses standards de cet anticoagulant. Plusieurs études rétrospectives sur des populations importantes de patients génotypés pour le CYP2C9 ont confirmé que les doses de warfarine, un autre coagulant coumarinique, nécessaires pour maintenir l’INR dans les limites souhaitées, diminuaient en présence d’un ou deux allèles mutés *2 et surtout *3 ce qui correspond au phénotype métaboliseur lent.
ROLE DES GENOTYPES DU CYP2C9 ET DE LA VITAMINE K EPOXYDE
REDUCTASE
La présence d’un allèle muté du
CYP2C9 *3 explique en partie la variabilité de la réponse au traitement anticoagulant par l’acenocoumarol mais d’autres facteurs génétiques sont potentiellement impliqués pour rendre compte de la variabilité de la réponse au traitement. En 2005, d’Andrea et coll . [17] mettent en évidence l’existence d’un polymorphisme génétique fonctionnel au niveau de l’enzyme vita-
mine K epoxyde réductase (VKORC1) expliquant également la variabilité de la réponse à la warfarine. Cet enzyme est le site d’action principal des anticoagulants.
Il permet dans le foie de régénérer à partir de la vitamine K oxydée la vitamine K réduite, cette dernière étant nécessaire à l’activation de la carboxylase vitamine K dépendante qui carboxyle les résidus glutamate et transforme ainsi en facteurs activables les facteurs de la coagulation synthétisés par le foie : prothrombine, proconvertine, facteur stuart et facteur anti-hemophilique B qu’on a appelé le complexe PPSB. Les anticoagulants oraux inhibent l’activité de la vitamine K epoxyde réductase. La présence d’un polymorphisme du promoteur du gène VKORC 1 (une adénine à la place d’une guanine en position 1639), entraîne une potentialisation de l’effet anticoagulant de la warfarine avec une diminution des doses nécessaires pour obtenir cet effet. Le SNP-1639 permet d’identifier avec certitude un haplotype particulier du gène VKORC1. Cet haplotype est une combinaison d’allèles spécifiques de plusieurs SNP repartis sur le gène VKORC1 dont quatre sont en complet déséquilibre de liaison. Cet haplotype particulier, plus simplement identifié par le SNP-1639, a été récemment associé à des niveaux d’expression hépatique de VKORC1 plus faibles que les autres haplotypes [18].
Cette baisse d’expression hépatique de VKORC1 en présence de l’haplotype particulier est vraisemblablement de nature transcriptionnelle par l’intermédiaire d’un allèle de l’haplotype dans le promoteur du gène qui diminue l’efficience de l’étape transcriptionnelle [17]. Ainsi la sensibilité accrue aux anticoagulants oraux détectée par le polymorphisme-1639 s’explique par une plus faible expression hépatique de la vitamine K époxyde réductase.
Nous avons donc effectué lors d’un quatrième travail, une étude haplotypique des différents SNP du gène VKORC 1 sur 222 échantillons d’ADN de l’étude précédente afin d’étudier différents polymorphismes et de les corréler à celui observé sur le CYP2C9 . L’objectif était de voir si l’association de deux polymorphismes génétiques (un sur le site récepteur, un sur l’enzyme du métabolisme) pouvait expliquer la variabilité de la réponse anticoagulante à l’acenocoumarol. Les résultats montrent que chez 25 %, de ces sujets la mutation du CYP2C9* 3 est associée à une mutation du gène VKORC 1 en position 1639 (une adénine à la place d’une guanine). La figure 1 montre la différence de l’effet anticoagulant (mesuré sur la variation du facteur VII et de l’INR) après une dose d’acenocoumarol en fonction de la présence d’un ou deux allèles mutés sur le gène VKORC 1.L’effet anticoagulant est plus important chez les sujets porteurs d’un ou deux variants allèliques du gène VKORC 1 que chez les sujets porteurs des allèles sauvages. Une analyse de régression multiple montre que la réponse au traitement anticoagulant est indépendamment corrélée à la présence de la mutation de l’enzyme CYP2C9 *3 et de la mutation du site effecteur de la vitamine K époxyde réductase. Lorsque ces deux mutations sont associées, elles expliquent plus de 50 % de la variabilité interindividuelle de la réponse au traitement [19]. La combinaison de ces deux variants allèliques fonctionnels sur deux gènes différents entraîne un risque dix fois plus élevé de surdosage en anticoagulant [20].
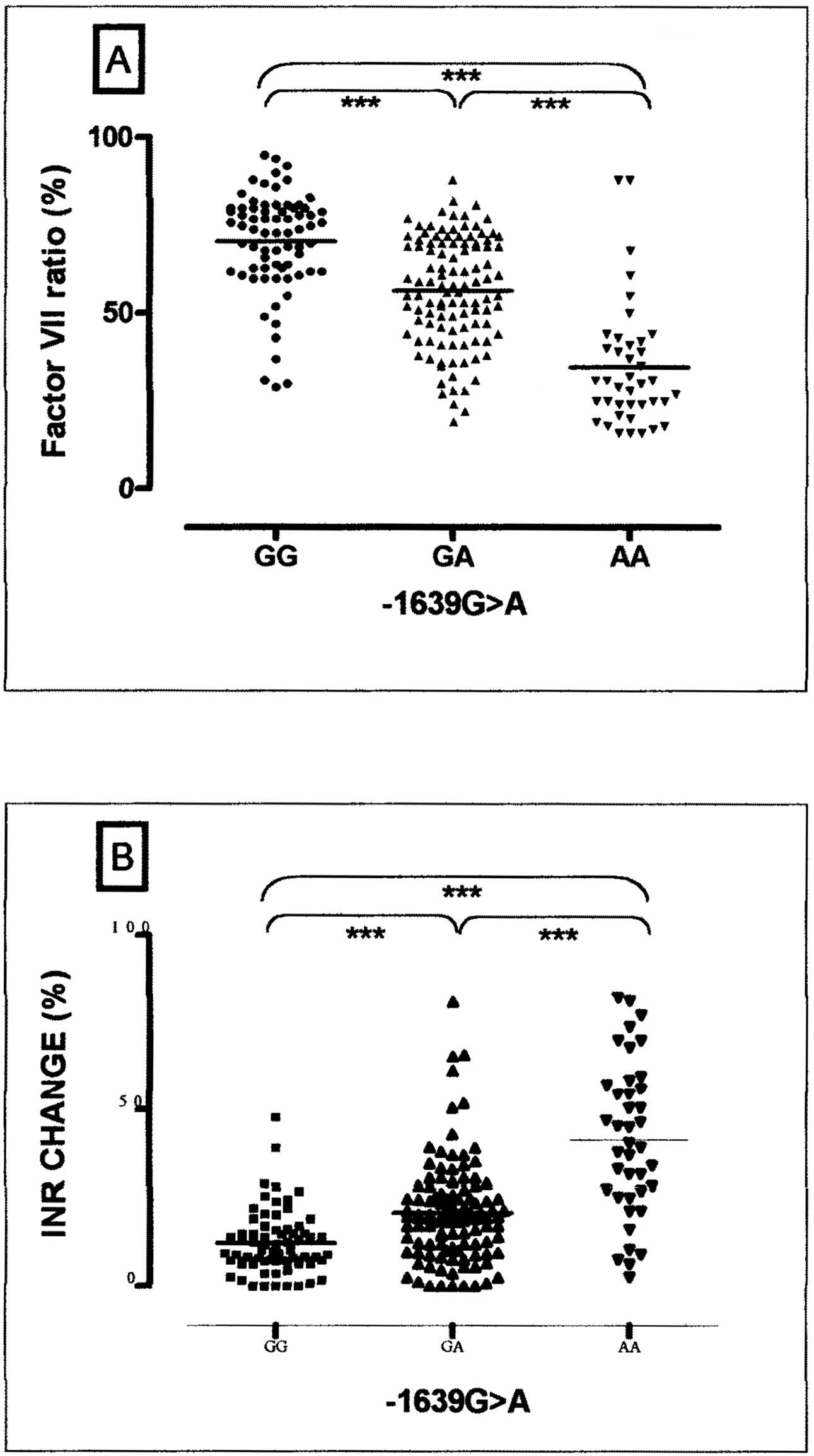
FIGURE 1 — Pourcentage de modification du facteur VII (A) et de l’INR (B) après l’absorption d’une dose unique d’acénocoumarol chez les sujets volontaires sains, en fonction du génotype de la vitamine K epoxyde reductase : GG génotype sauvage homozygote, GA hétérozygote, AA homozygotes mutés (222 sujets volontaires sains) (xxx = p. < 0,001) [ref.18]. (Figure originale publiée dans Blood. BODIN L., VERSTUYFT C., TREGOUET D.A., ROBERT A., DUBERT L., FUNCK-BRETANO C., JAILLON P., BEAUNE P., LAURENT-PUIG P., BECQUEMONT L., LORIOT M.A., — Cytochrome P450 2C9 (CYP2C9) and vitamin K epoxyde reductase (VKORC1) genotypes as determinants of acenocoumarol sensitivity — Blood, 2005, 106, 135- 140 © the American Society of Hematology).
Ces résultats montrent que l’étude simultanée du génotype de l’enzyme de métabolisation et du génotype du site effecteur des anticoagulants coumariniques permet de prédire avec une plus grande précision la réponse au traitement chez un individu donné. Les métaboliseurs lents homozygotes CYP2C9 *3/*3 devraient recevoir une dose beaucoup plus faible de warfarine ou d’acenocoumarol s’ils sont porteurs, en plus, d’une mutation génétique sur le gène de la vitamine K-epoxyde réductase. En effet ces génotypes sont prédictifs d’un risque de surdosage en anticoagulant accompagné d’un risque hémorragique important, notamment en début de traitement. Il est possible enfin que le risque d’interactions médicamenteuses entre les anticoagulants coumariniques et les anti-inflammatoires non stéroïdiens soit augmenté chez les sujets porteurs de l’allèle muté CYP2C9 *3 comme cela a été récemment suggéré [21]. Des essais cliniques prospectifs devraient maintenant explorer ces questions.
PROSPECTIVE
Les travaux que nous venons de résumer illustrent les trois étapes principales de la recherche en Pharmacogénétique aujourd’hui. Si l’on veut que ces recherches aboutissent à des recommandations pratiques applicables par les praticiens, il faut :
— identifier des médicaments à ‘‘ haut risque ’’, c’est-à-dire ceux dont les concentrations toxiques sont proches des concentrations thérapeutiques dans la pratique courante, ce qui limite grandement les possibilités d’adaptation posologique pour le praticien. De plus, si la toxicité de ces médicaments peut mettre en jeu le pronostic vital (comme c’est le cas pour les anticoagulants), il est parfaitement justifié d’essayer de prévoir à l’avance l’adaptation de la posologie en fonction des caractéristiques génétiques des individus en réalisant précocement un test génétique simple.
— identifier pour ces médicaments les voies métaboliques chargées de leur élimination chez l’homme et savoir s’il existe, dans une population donnée, un polymorphisme génétique des enzymes qui sont impliquées dans ce métabolisme.
— enfin, étudier les conséquences de ce polymorphisme génétique, s’il existe, sur la cinétique d’élimination du médicament et sur l’intensité de ses effets pharmacodynamiques ou thérapeutiques ou de ses effets potentiellement toxiques dans une population importante.
Ces études permettront d’évaluer chez l’homme le rôle de ces génotypes dans la variabilité de la réponse aux traitements. Les conséquences d’une association de plusieurs génotypes mutés concernant non seulement les enzymes du métabolisme, les systèmes transporteurs transmembranaires des médicaments, et les récepteurs ou sites effecteurs des médicaments doivent être systématiquement recherchées.
Ces recherches en Pharmacogénétique permettront-elles un jour d’atteindre l’objectif d’une adaptation individuelle de la posologie de certains médicaments en fonction du code génétique ? Nous le pensons mais le chemin à parcourir est encore long ! Aujourd’hui, les praticiens ont déjà appris à adapter leur prescription en
fonction du génotype de certains enzymes du métabolisme. L’exemple le plus connu et le plus significatif est celui de la prescription d’immunosuppresseurs, azathioprine (IMURELR) ou mercaptopurine chez les enfants atteints de leucémie hymphoblastique aiguë ou chez les patients atteints de maladie de Crohn. Un déficit de l’enzyme thiopurine-S- methyltransférase TPMT chez certains sujets homozygotes entraîne un risque majeur de neutropénie grave due à l’accumulation de métabolites toxiques, les 6 thioguanine nucléotides. Les séquences du gène de la TPMT, sièges de mutations ont été identifiées et l’allèle TPMT *3A le plus fréquemment responsable d’un phénotype métaboliseur lent est présent chez 4 % des sujets caucasiens [22]. Les sujets hétérozygotes représentent 6 à 11 % des caucasiens et leur enzyme TPMT présente une activité intermédiaire entre celle des sujets homozygotes sauvages et celle des sujets homozygotes mutés où elle est très faible (0,2 à 0,6 % de la population caucasienne) [23]. Chez les sujets homozygotes mutés les doses d’immunosuppresseurs doivent être considérablement diminuées à cause du risque de toxicité hématologique majeure.
Nous pensons que les recherches sur la pharmacogénétique des récepteurs comme celles consacrées aux récepteurs béta-adrénergiques dans l’insuffisance cardiaque [24], déboucheront un jour sur une meilleure compréhension des processus pathologiques et une possibilité d’adaptation thérapeutique individuelle. Cependant pour que les tests pharmacogénétiques entrent dans la pratique quotidienne des médecins, il faut que soient remplies les trois conditions suivantes :
— disposer d’un test génétique facile à réaliser à moindre coût sur l’ADN génomique de cellules faciles à prélever (leucocytes circulants) ce qui est déjà le cas aujourd’hui — avoir démontré chez l’homme les valeurs prédictives positives et négatives du test génétique et l’intérêt du test dans l’adaptation posologique individuelle — avoir montré dans des essais cliniques prospectifs que la réalisation du test génétique, menant à une adaptation posologique individuelle, apporte un béné- fice pour le patient et diminue le risque d’effets indésirables ou d’interactions médicamenteuses.
CONCLUSION
En prenant appui sur les travaux de recherche clinique effectués dans notre équipe, nous avons voulu illustrer la progression de l’exploration des retombées cliniques de la pharmacogénétique. En prenant comme sujet d’observation un anticoagulant, l’acenocoumarol (SINTROM®) nous avons montré que chez certains patients, la présence de variants allèliques du CYP2C9 principal enzyme du métabolisme de ce médicament, correspondant à un phénotype métaboliseur lent, entraînait un risque de surdosage et un risque hémorragique. En associant l’étude de mutations sur l’enzyme responsable du métabolisme et de mutations sur le site récepteur de ce médicament, nous avons montré que la prédiction de la réponse au traitement
augmentait considérablement et que cela devait permettre, chez les patients porteurs de ces mutations, de diminuer les doses d’acenocoumarol afin de réduire le risque de complications hémorragiques liées au traitement. Ces études de pharmacologie clinique doivent maintenant déboucher sur des essais cliniques thérapeutiques prospectifs qui permettront d’évaluer l’intérêt pratique de ces tests génétiques dans des populations importantes de patients et de quantifier la diminution du risque iatrogène médicamenteux.
Nous ne sommes aujourd’hui qu’aux prolégomènes de cette nouvelle science médicale qu’est la pharmacogénétique. Mais ce qui est clair déjà actuellement, c’est qu’on ne peut plus explorer la pharmacocinétique et les propriétés pharmacodynamiques d’un nouveau médicament chez l’homme sans tenir compte d’éventuels facteurs génétiques qui expliqueraient la variabilité de la réponse individuelle au traitement. Ces facteurs génétiques concernent principalement les enzymes du métabolisme des médicaments, les systèmes transporteurs transmembranaires et les sites récepteurs de ces médicaments. Le développement de la pharmacogénétique et de la pharmacogénomique ne fait que commencer.
BIBLIOGRAPHIE [1] BONICKE R., REIF W. — Enzymatische inaklivierung von isonicotinsaure hydrazide im meuschlichen und tierischen organismus — Arch. Exp. Pathol. Pharmakol., 1953 , 220, 321-33 .
[2] HUGHES H.B, BIEHL J.P., JONES A.P, SCHMIDT L.H. — On the metabolic fate of isoniazid
J.
Pharmacol Exp. Therap , 1953 , 109, 444-452 .
[3] HUGHES H.B., BIEHL J.P., JONES A.P., SCHMIDT LH. — Metabolism of isoniazid in man as related to the occurrence of peripheral neuritis — Am. Rev. Tuberc, 1954 , 70, 266-273.
[4] CARSON P.E., FLANAGAN C.L, ICKES C.E., ALVONG A.S. — Enzymatic deficiency in primaquine sensitive erythrocytes, Science , 1956 , 124, 484-485.
[5] KALOW W., GENEST K. — A method for the detection of atypical forms of human serum cholinesterase. Determination of dibucaine numbers, Can. K. Biochem. Physiol., 1957 , 35, 339-346.
[6] NEBERT D.W., RUSSELL D.W. — Clinical importance of the cytochromes P450 —
Lancet, 2002 , 360, 1155-1162.
[7] LEE C.R., GOLDSTEIN J.A., PIEPER J.A — Cytochrome P450 2C9 phymorphisms : a comprehensive review of the in-vitro and human data — Pharmacogenetics , 2002 , 12, 251-263.
[8] YANG J.Q., MORIN S., VERSTUYFT C., FAN L.A., ZHANG Y., XU C.D., BARBU V. et al. —
Frequency of cytochrome P450 2C9 allelic variants in the chinese and french populations
Fund.
Clin. Pharmacol., 2003, 17, 373-376.
[9] PEDERSEN R.S., VERSTUYFT C., BECQUEMONT L., JAILLON P., BROSEN K. — Cytochrome P450 2C9 (CYP2C9) genotypes in a nordic population in Denmark — Basic Clin. Pharmacol.
Toxicol., 2004, 94, 151-152.
[10] THIJSSEN H., FLINOIS J.P, BEAUNE P. — Cytochrome P450 2C9 is the principal catalyst of racemic acenocoumarol hydroxylation reactions in human liver microsomes — Drug Metab. Dispos., 2000 , 28, 1284 – 1290.
[11] STEWARD D.J., HAINING R.L., HENNE K.R., DAVIS G., RUSHMORE T.H., TRAGER W.F. et al. —
Genetic association between sensitivity to warfarin and expression of CYP2C9*3 —
Pharmacogenetics, 1997, 7, 361-367.
[12] THIJSSEN H., VERKOOIJEN I., FRANK H. — The possession of the CYP 2C9*3 allele is associated with low dose requirement of acenocoumarol — Pharmacogenetics , 2000 , 10, 757-760.
[13] OGG M.S., BRENNAN P., MEADE T., HUMPHRIES S.E. — CYP 2C9*3 allelic variant and bleeding complications. Lancet, 1999, 354 , 1124.
[14] VERSTUYFT C., MORIN S., ROBERT A., LORIOT M.A., BEAUNE P., JAILLON P. et al. — Early acenocoumarol overanticoagulation among cytochrome P450 2C9 poor metabolisers.
Pharmacogenetics, 2001 , 11 , 735-737.
[15] VERSTUYFT C., ROBERT A., MORIN S., LORIOT M.A, FLAHAUT A., BEAUNE P., et al. — Genetic and environmental risk factors for oral anticoagulant overdose —
Eur. J. Clin. Pharmacol., 2003 , 58, 739-745.
[16] MORIN S., BODIN L., LORIOT M.A., THIJSSEN H.H.W., ROBERT A., STRABACH S. et al. —
Pharmacogenetics of acenocoumarol pharmacodynamics —
Clin. Pharmacol. Ther., 2004 , 75, 403-414.
[17] D’ANDREA G., D’AMBRIOSO R.L., DI PERNA P., CHETTA M., SANTACROCE R. et al. — A polymorphism in the VKOR C1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood, 2005 , 105, 645-649.
[18]
R IEDER M.J., REINER A.P., GAGA B.F., NICKERSON D.A., EBY C.S., MCLEOD H.L. et al. — Effect of VKORC1 Haplotypes ou transcriptional regulation and warfarin dose.
NEJM , 2005, 352 , 2285-2293.
[19] BODIN L., VERSTUYFT C., TREGOUET D.A., ROBERT A., DUBERT L., FUNCK-BRENTANO C. et al.
— Cytochrome P450 2C9 (CYP2C9) and vitamin K epoxyde reductase (VKORC1) genotypes as determinants of acenocoumarol sensitivity. Blood, 2005 , 106, 135-140.
[20] OUTEINEH L., VERSTUYFT C., DESCOT C., DUBERT L., ROBERT A., JAILLON P. et al. — Vitamin K epoxyde reductase (VKORC) genetic polymorphism is associated to oral anticoagulant overdose. Thromb. Haemost., 2005, 94 , 690-691.
[21] VISSER L.E., VAN SHAIK R., VAN VLIET M., TRIENEKENS P.H., DESMET P.A., VULTO AG. et al. —
Allelic variants of cytochrome P450 2C9 modify the interaction between nonsteroidal antiinflammatory drugs and coumarin anticoagulants. Clin. Pharmacol. Ther., 2005 , 77, 479- 485.
[22] LENNARD L., VAN LOON J.A., WEINSHILBOUM R.M. — Pharmacogenetics of acute azathioprine toxicity : relationship to thiopurine methyltransferase genetic polymorphism. Clin. Pharmacol.
Ther., 1989 , 46, 149-154.
[23] STANULLA M., SCHAEFFELER E., FLOHR T., CARLO G., SCHRAUDER A., ZIMMERMANN M. et al . —
Thiopurine methyltransferase (TMPT) genotype and early treatment response to mercaptopurine in childhood acute hymphoblastic lenkenia. JAMA, 2005 , 293, 1485-1489.
[24] TERRA S.G., PAULY D.F., LEE C.R., PATTERSON J.H., ADAMS K.F., SCHOFIELD R.S. et al — β — adrenergic receptor polymorphisms and responses during titration of metoprohol controlled release / extended release in heart failure. Clin. Pharmacol. Ther., 2005 , 77, 127-137.
DISCUSSION
M. Roger BOULU
Avez-vous des précisions sur la réalisation des tests ?
Ces tests biologiques de pharmacogénétique sont maintenant aisément praticables. Les techniques utilisant la PCR en temps réel avec des sondes fluorescentes sont très reproductibles, de coût de plus en plus réduit et surtout très rapides (environ deux heures). Par ailleurs, des puces à génotypages avec marquage CE ont récemment été mises en vente pour le génotypage des CYP2D6 et CYP2C9. Enfin, il est fort probable que d’ici quelques années, quand l’intérêt clinique des principaux polymorphismes aura été précisé, que nous disposions de puces à génotypage qui puissent être utilisées dès la naissance comme on dépiste actuellement l’hypothyroïdie ; les données figureront ensuite sur la carte de santé de chaque assuré social à la disposition des praticiens.
M. Alain LARCAN
En pratique dans la surveillance du traitement anticoagulant : hypersensibilité ou résistance, on commence à rechercher des facteurs liés à l’alimentation ou à une affection hépatique méconnue (alcoolisme) et surtout à des associations médicamenteuses (induction enzymatique) avec des effets potentialisateurs des AINS, par exemple des antagonistes des anticomitiaux. Comment réagissent les porteurs d’anomalies génomiques à ces facteurs associés ?
Existe-t-il des différences selon la famille des anti-vitamine K, coumariniques et phénylindanédione ?
Il existe naturellement de fortes interactions génome-environnement. Les phénomènes d’interactions médicamenteuses seront d’autant plus prononcés que les sujets sont déjà génétiquement plus sensibles à l’action des AVK du fait de leur polymorphisme VKORC1 ou de leur déficience en élimination métabolique CYP2C9- dépendante. Il en va de même avec les pathologies associées sous jacentes comme les cirrhoses ou les hépatites chroniques actives. En ce qui concerne les deux différentes classes d’anticoagulants oraux (coumariniques et indanes diones) les conséquences du polymorphisme VKORC1 sont strictement superposables. Il est assez probable, à la lumière de recherches actuellement en cours dans l’équipe de Patrice Jaillon à SaintAntoine que le CYP2C9 soit aussi impliqué dans l’élimination de la fluindione (Previscan®) mais dans une moindre mesure que pour les dérivés coumariniques (acenocoumarol et warfarine).
M. Pierre GODEAU
La pharmacogénétique explique-t-elle certaines interactions médicamenteuses nouvellement constatées dans l’utilisation des AVK. Trois exemples : le paracétamol, les inhibiteurs de la réception de la sérotonine, les glycocorticoïdes ?
Non, ou tout du moins pas de façon prépondérante. En ce qui concerne le paracétamol, c’est un de ses métabolites qui vient inhiber la vitamine K époxyde réductase comme récemment démontré par Thijssen et col ( Thromb. Haemost. 2004, Oct) 92(4) 797-802).
Cette inhibition de la vitamine K époxyde réductase vient s’ajouter à celle conférée par les anticoagulants oraux et il en résulte une interaction purement pharmacodynamique au niveau de la cible d’action des AVK. Il est possible que l’amplitude de l’interaction soit modulée par le polymorphisme génétique de VKORC1 mais cela reste à démontrer. En ce qui concerne les interactions avec les inhibiteurs de recapture de la sérotonine, il s’agit en revanche d’une interaction médicamenteuse d’ordre pharmacocinétique : les IRSS sont pour la plupart des inhibiteurs plus ou moins puissants du CYP2C9. Ils ralentissent l’élimination des AVK, les concentrations circulantes de ces derniers s’élèvent ainsi que l’INR. Il est probable que les patients partiellement déficients en CYP2C9 du fait du polymorphisme génétique de ce gène soit plus exposés aux interactions médicamenteuses avec les IRSS ou les autres inhibiteurs du CYP2C9 mais cela reste encore à démontrer.
Enfin, pour les glucocorticoïdes, l’interaction qu’ils génèrent, utilisés à fortes doses avec les AVK, reste à ma connaissance sans explication.
M. Bernard LAUNOIS
L’Azathioprine est un vieil immunosuppresseur. On est étonné qu’il y ait eu un accident mortel car il s’agit d’un médicament ancien, peu efficace. S’agit-il d’expérience ancienne ou récente.
Bien que l’azathioprine soit un « vieil immunosuppresseur », il est néanmoins largement utilisé dans certaines leucémies de l’enfant et il s’est imposé dans les dix dernières années comme un traitement de référence de la maladie de Crohn résistante aux glucocorticoï- des, indications dans lesquelles il est toujours considéré comme remarquablement efficace. Les neutropénies graves décrites avec l’azathioprine sont pour l’essentiel des cas des descriptions anciennes, mais cela tient principalement au fait que le génotypage de la TPMT est devenu en Occident à peu prêt systématique avant l’introduction de ce médicament, permettant de proposer aux patients métaboliseurs lents soit un traitement alternatif (anti TNF dans la maladie de Crohn) ou une réduction de posologie très importante avec une surveillance hématologique rapprochée dans les leucémies.
M. Jean-Daniel SRAER
Est-il nécessaire de faire des tests de pharmacogénomique, en dehors d’études scientifiques, pour tous les immunosuppresseurs ? Les interactions médicamenteuses ne semblent-elles pas plus importantes ainsi que les conditions environnementales ou alimentaires ?
Vous avez parfaitement raison de souligner que l’essentiel de la variabilité interindividuelle de réponse aux médicaments est d’ordre environnementale et non génétique.
Notre propos n’est pas d’imposer la pharmacogénétique comme critère explicatif principal, mais de contribuer à diffuser l’information auprès des médecins sur les aspects génétiques de la variabilité de réponse aux médicaments, dans les cas où l’influence génétique sur la réponse aux traitement a été démontrée et ce de façon cliniquement pertinente. Ainsi, pour le tacrolimus en transplantation rénale, il semble préférable de pratiquer un génotypage du CYP3A5, impliqué dans le métabolisme de cet immunosuppresseur chez les patients inscrits sur la liste d’attente de greffe afin de mieux prévoir la
dose initiale qui doit être proposée, en attendant d’adapter cette dernière en fonction des taux circulants dans les premiers jours de la greffe. En général, les tests pharmacogéné- tiques sont utiles dans la période d’introduction du traitement soit pour anticiper sur la posologie la plus adaptée au patient, soit pour caractériser le patient comme potentiellement répondeur ou non ou encore comme à risque de développer un événement indésirable. Toute variation de réponse à un médicament survenant après une longue exposition à un même traitement doit être prioritairement attribuée à un facteur environnemental et non à un facteur génétique.
M. Roger NORDMANN
Vous avez parfaitement explicité la relation entre le polymorphisme génétique (portant notamment sur le cytochrome P450 2C9) et la réponse aux anti-vitamines K. Cette réponse dépend cependant de l’activité de l’enzyme au moment même de la prise de l’anti-vitamine.
Les cytochromes P450 étant généralement inductibles, il doit en être de même pour le 2C9 et il est vraisemblable que la réponse au traitement dépend de son activité éventuellement induite soit par le médicament lui-même, soit par d’autres facteurs (tels que le tabac et/ou l’alcool). A-t-on observé une différence dans le degré d’inductibilité selon que le génotype correspond à des métaboliseurs lents ou rapides ?
La encore, il s’agit d’une très intéressante question sur les relation génome — environnement. En effet, le CYP2C9 est inductible comme d’autres CYP par des médicaments comme la rifampicine ou les anticonvulsivants de première génération ou encore des plantes comme le Millepertuis (St John’s wort). Jusqu’à présent, personne n’a pu mettre en évidence de contrôle génétique de l’amplitude de l’induction du CYP2C9. Pour le CYP1A2, inductible par les produits de combustion du tabac notamment, l’amplitude d’induction de cet enzyme est en partie dépendante d’un polymorphisme génétique dans le promoteur du gène séparant ainsi les patients exposés au tabac en » fort inducteurs » et « faibles inducteurs ».
M. Patrice QUENEAU
Concernant les relais héparine-antivatimine K, doit-on entrer dans une pratique d’étude du génotype chez les malades à risques ? Et ce d’autant que les surcoûts liés aux tests devraient être largement compensés par l’épargne des complications et de leurs surcoûts. Qu’en pensez-vous ? Pour prendre un autre exemple, celui des AINS, peut-on considérer, dès lors, comme dès à présent souhaitable, voire nécessaire de réaliser ces tests chez les personnes à risque, par exemple chez les personnes âgées justiciables de traitements prolongés par les AINS, de même bien sûr que chez les malades justiciables d’un traitement par AINS malgré des antécédents d’hémorragie digestive ; et ce, en tenant compte en outre des influences génotypiques telles que vous les avez évoquées concernant les inhibiteurs de la pompe à protons.
En ce qui concerne l’instauration systématique d’un génotypage avant l’instauration d’un traitement par AVK, je pense en effet que les données actuelles sont suffisantes pour le proposer de facon systématique. La FDA réfléchi de son coté à intégrer dans les recommandations des AVK (équivalemnt de nos RCP du Vidal) la pratique d’un génotypage CYP2C9 et VKORC1. Il manque cependant encore une étude randomisée démontrant que le génotypage avant l’introduction des AVK permet de diminuer les
accidents hémorragiques en début de traitement. Pour la protection gastrique par inhibiteurs de la pompe à proton (IPP) chez les patients traités par AINS, nous ne disposons pas des preuves suffisantes pour demander la généralisation d’un génotypage du CYP2C19. En effet, les données pharmacogénétiques portent non pas sur la prévention mais sur le traitement des ulcères gastro duodénaux secondaires à Helico Bacter Pylori. De plus, les données publiées indiquent que chez les métaboliseurs rapides du CYP2C19, le taux de guérison est d’environ 80 % au décours d’une trithérapie avec de l’oméprazole dosé à 20 mg / j. Les recommandations actuelles du traitement de ces affections indiquent une posologie double, soit 40 mg/j ; il est fort probable qu’avec 40mg par jour d’oméprazole, le taux de guérison chez les métaboliseurs rapides du CYP2C19 soit bien plus élevé, rendant moins utile le génotypage CYP2C19 à la recherche de « mauvais répondeurs ».
* Service de Pharmacologie, Faculté de Médecine Pierre et Marie Curie (St-Antoine)-Université Paris 6 et Assistance Publique-Hopitaux de Paris ** Laboratoire de Génétique Moléculaire et Pharmacogénétique, CIB Paris-sud Faculté de Médecine de Paris-sud, Université Paris XI et Assistance Publique-Hôpitaux de Paris. Tirés-à-part : Professeur Laurent Becquemont, même adresse. Article reçu et accepté le 16 janvier 2006
Bull. Acad. Natle Méd., 2006, 190, no 1, 37-53, séance du 3 janvier 2006