
Résumé
La dystrophie facio scapulo humérale (DMFSH) est une des myopathies les plus fréquentes. Son nom est directement lié à une répartition particulière de l’atteinte musculaire, souvent asymétrique, qui, pendant une grande partie de l’évolution, reste limitée à certains territoires laissant coexister chez le même patient des muscles très déficitaires et des muscles non atteints. Lui est associée une mutation particulière faite de la délétion d’un nombre entier de séquence répétée, appelée D4Z4, située sur la région télomérique du chromosome 4. Aucune thérapeutique n’est actuellement disponible. Une thérapie génique reste inenvisageable puisque aucune altération n’a été objectivée dans les gènes localisés dans la région incriminée. D’un autre coté, s’agissant d’une dystrophie musculaire une des caractéristiques est la coexistence d’une dégénérescence du tissu musculaire génétiquement induite, longtemps compensée par un mécanisme de régénérescence du muscle à partir des cellules satellites, et ce n’est que lorsque ce mécanisme est dépassé que se manifestent déficience et atrophie. Dans ce contexte, une thérapie cellulaire utilisant des myoblastes produits à partir de cellules satellites du patient lui-même devrait permettre de renforcer les capacités régénératrices du muscle et protéger, même si cela reste temporaire, contre la dégénérescence et ses conséquences fonctionnelles. Cela éviterait également l’utilisation de thérapies immunosuppressives potentiellement toxiques. Pour tester cette hypothèse de travail nous avons étudié, ex vivo et in vivo sur des souris immunodéficientes les capacités de régénération et de différenciation de cellules satellites de patients atteints de DMFSH en utilisant une méthodologie de culture à haut rendement récemment développée. Une biopsie musculaire a été réalisée chez 5 patients, après consentement informé. Leur présentation clinique répondait aux critères diagnostiques de la maladie et tous avaient une délétion D4Z4 significative. La biopsie fut réalisée dans un muscle vaste latéral où il n’existait aucun signe d’atteinte dystrophique, clinique, radiologique et anatomo-pathologique. Les caractéristiques de croissance incluant calcul des rendements et l’expression du phénotype sur marquage CD56 et desmine ainsi que les capacités de myocytes exprimant la dystrophine humaine chez les souris immunodéficientes Rag2 ont été analysées et comparées à des muscles témoins. Les rendements et temps de prolifération à partir de 1 g de tissu prélevé étaient comparables aux témoins ainsi que les capacités à produire des myotubes et des cellules musculaires différenciées. Cette étude montre que, si les cellules satellites sont obtenues à partir d’un muscle non dystrophique chez un sujet atteint de DMFSH, les qualités des myoblates produits permettent leur utilisation dans un essai de thérapie cellulaire chez l’homme.
Summary
Facioscapulohumeral dystrophy (FSHD), one of the most common forms of muscular dystrophy, derives its name from the patients’ selective, often asymmetric clinical distribution of muscle weakness. Interestingly, affected and non affected areas can coexist in the same patient for many years. The molecular hallmark is total deletion of the subtelomeric D4Z4 repeat on chromosome 4q. There is no specific treatment. Gene therapy is unlikely to be feasible, as no alterations have been found in the genes located in this subtelomeric region. Muscular dystrophies are characterized by the coexistence of genetically induced muscle degeneration and compensatory muscle regeneration by myoblast proliferation from satellite cells ; muscle weakness and atrophy appears when this mechanism is overwhelmed. Cell therapy with autologous myoblasts would, in theory, be a simple way of boosting the regenerative process and of preventing or delaying muscle degeneration. This approach might also avoid the use of toxic immunotherapies. By using a recent very-high-yield cell culture method, we analyzed the proliferation and differentiation of myoblasts obtained from FSHD patients, both ex vivo and in vivo (by intramuscular injection to immunodeficient mice). Myoblasts were obtained by muscle biopsy from five FSHD patients harboring the D4Z4 deletion. We selected the vastus lateralis muscle, which exhibited no clinical, radiological or pathological signs of dystrophy. The growth characteristics of these cells were compared with those of cells from normal control muscles, based on the culture yield, phenotypic characterization with anti-CD56 and anti-desmin antibodies, and the capacity for differentiation (myotube production in vitro and human dystrophin expression one month after injection to Rag2 immunodeficient mice). Patients’ cells recovered from 1 g of muscle biopsy specimen resembled control cells in terms of their growth kinetics, culture yield, and capacity to differentiate and produce mature muscle cells. These results indicate that myoblasts taken from unaffected muscle of patients with FSHD warrant testing in a human cell therapy trial.
INTRODUCTION
La dystrophie musculaire facio-scapulo-humérale (DMFSH) est une des myopathies héréditaire les plus fréquentes (prévalence d’environ 1 sur 20.000) [1, 2]. Sa transmission est autosomique dominante liée à la délétion d’un nombre entier de copies d’une séquence répétée de 3,3 Kb appelée D4Z4 localisée dans la région télomérique du bras long du chromosome 4, ne laissant qu’un fragment inférieur à 35 Kb, alors que chez le sujet normal la taille de cette séquence varie de 50 à 300 Kb [3 ,4]. La taille de la délétion serait corrélée à l’âge d’apparition de la maladie et à la sévérité du tableau clinique [5] mais son rôle physiopathologique reste inconnu. Une hypothèse récente serait que la délétion permette une modification structurale du fragment d’ADN concerné permettant une dérépression transcriptionnelle de gènes normalement non exprimés et notamment du gène FRG1 [6, 7].
Cliniquement la DMFSH se caractérise par une atteinte de topographie spécifique dont elle tire son nom, souvent asymétrique, touchant toujours les muscles fixateurs de l’omoplate, les muscles scapulaires, de l’avant-bras et, chez la majorité des sujets la face. À un stade plus avancé les muscles d’abord jambiers antérieurs puis quadricipitaux et ischiojambiers sont touchés et enfin les muscles des bras. L’évolution est très lentement progressive et de ce fait, de nombreux patients présentent longtemps la co-existence de muscles phénotypiquement normaux et de muscles déficitaires et atrophiques. Il n’y a généralement aucune autre atteinte, rarement une surdité, une atteinte rétinienne, un retard mental ou une épilepsie.
La DMFSH fait partie du groupe des dystrophies musculaires progressives selon le concept développé par Erb dès 1891. Ces dystrophies musculaires correspondent à une dégénérescence fibro-graisseuse du tissu musculaire d’évolution progressive, génétiquement induite, touchant des muscles différenciés, dépassant progressivement le mécanisme de restauration physiologique par différenciation des cellules satellites. Les cellules satellites sont en effet capables, en réponse à la surexpression de facteurs de régulation myogénique, de donner des myoblastes régénérateurs qui progressivement fusionnent pour former des myotubes tandis que myofilaments et myofibrilles se développent [8]. Selon le type de dystrophie musculaire et son génie évolutif, ces mécanismes de régénération compensent plus ou moins longtemps la dégénérescence musculaire génétiquement induite, la perte fonctionnelle musculaire squelettique et la déficience motrice. Les handicaps conséquents n’apparaissent que quand le mécanisme de régénération est dépassé [9].
De ce fait, dans la mesure où à ce jour aucun moyen de thérapie pharmacologique n’est disponible ou de thérapie génique n’est envisageable pour compenser cette déficience, une thérapie cellulaire avec apport de myoblastes capable de renforcer focalement la régénérescence pourrait permettre de prolonger les capacités de maintien du tissu musculaire et ainsi, de façon symptomatique, compenser même temporairement, la dégénérescence et la perte cellulaire. La DMFSH représente un modèle d’autant plus séduisant qu’elle est caractérisée par la co-existence
longtemps conservée de zones fonctionnellement saines et d’autres déficitaires. C’est pourquoi, et dans le but de contourner les difficultés liées au rejet de greffes et aux traitements immunosuppresseurs au long court, nous nous intéressons particuliè- rement à la possibilité d’une thérapie cellulaire autologue dans cette pathologie. Ce concept, utilisant des cellules porteuses de l’anomalie génétique, ne peut avoir un but curatif mais a pour objectif de prolonger la fonction motrice en apportant localement des éléments de maintien de la structure fonctionnelle. En préliminaire nous avons réalisé une étude des capacités prolifératives et régénératives des myoblastes obtenus à partir de patients atteints de DMFSH afin de juger de la possibilité de leur utilisation à des fins thérapeutiques. Nous présentons les résultats de cette étude.
MATÉRIELS ET MÉTHODES
Les patients
Cinq patients atteints DMFSH ont été sélectionnés selon des critères d’inclusion définis. Chaque patient, après consentement informé, a eu un prélèvement d’environ 1 g de muscle par biopsie ouverte selon les techniques habituelles utilisées en pratique diagnostique. Déposé sur une compresse stérile le prélèvement était ensuite directement plongé dans un milieu de transfert additionné d’antibiotiques à tempé- rature ambiante et transporté entre 4 et 8° C en container isotherme vers le laboratoire.
Les caractéristiques des patients sont données dans le tableau I. Les témoins contrôles sont des patients ayant fourni une biopsie musculaire dans le cadre d’une étude réalisée pour l’étude de l’implantation de myoblastes dans la cicatrice de l’infarctus du myocarde, ne présentant pas de dystrophie musculaire. Les moyennes d’âge sont semblables et les prélèvements effectués sur le muscle vaste externe comme pour les sujets dystrophiques.
Production cellulaire
La mise en culture et l’expansion ont été réalisées selon les procédures standard utilisées au Laboratoire de Thérapie Cellulaire de l’Hôpital Saint Louis [10] selon une technique validée (Vilquin et al Brevet no 007304, licence Société Myosix SA). La technique de production fait appel à l’éminçage fin du tissu musculaire, à la digestion puis à des séries d’expansions réalisées en présence d’un milieu de croissance sélectif. Après récolte les cellules ont été conservées dans un milieu de survie adéquat. Les cellules ont pu être congelées de la même façon que les produits sanguins traditionnels en présence d’albumine humaine et de diméthylsulfixide.
TABLEAU I. — Récapitulatif des caractéristiques des patients DMFSH biopsies (M = masculin, VE G = muscle vaste externe gauche). Le nombre de répétition D4Z4 caractérise la délétion en 4q35, tous les patients ayant un phénotype caractéristique de la maladie, le nombre de 10 répétitions est retenu comme marqueur moléculaire génétique significatif de DMFSH. Le stade fonctionnel de Brooke et Vignos (Vignos et al. JAMA 1963,184 :89-96) est utilisé en routine clinique pour exprimer les capacités fonctionnelles sur une échelle de 6 points croissant avec la déficience pour les membres supérieurs (1er chiffre) et de 10 points croissant avec la déficience pour les membres inférieurs (2e chiffre), le ratio donné pour tous les patients montre ici qu’ils sont à un stade fonctionnel identique et qu’il n’y a pas de déficience fonctionnelle dans les membres inférieurs : Le gradation IRM est faite selon une échelle de 3 valeurs appliquée ici aux loges quadricipitales sur séquence pondérée en T1 : I = absence d’hypersignaux graisseux, II = hypersignaux graisseux dans le muscle, III = remplacement du signal musculaire par un signal graisseux.
Repet.
Stade Ftnel
IRM
Poids sec
No
Sexe
Âge
Biopsie
D4Z4
Brooke grade (grammes)
FSH01 M 45 10 2/1 I VE G 1,15 FSH02 M 53 6 2/1 I VE G 1,12 FSH03 M 59 9 2/1 I VE G 1,05 FSH04 M 51 6 2/1 I VE G 0,91 FSH05 M 55 7 2/1 I VE G 0,98 Caractérisation in vitro
Numération cellulaire et rendement théorique
Les numérations cellulaires ont été réalisées à l’aide de cellules de comptage manuel sur lames de Malassez ; comptages réalisés deux fois par deux observateurs indé- pendants. Ces numérations, réalisées à toutes les étapes de la production, ont permis d’évaluer les rendements théoriques établis en repiquant successivement en boîtes de culture, au-delà du 5ème ou du 6ème passages les cellules en culture permettant d’obtenir un rapport de prolifération. Le rendement final est obtenu en multipliant entre eux les rapports de prolifération intermédiaire.
Caractérisation phénotypique par cytométrie de flux
CD 56 (Cluster differenciation 56) est une molécule d’adhésion de type NCAM exprimée dans les cellules satellites, les myoblastes, les myotubes et les fibres musculaires en régénération [11]. L’anticorps anti-CD56 est dirigé contre la partie extracellulaire de la protéine ce qui permet l’utilisation de fluorophore de flux au phosphore pour la repérer ainsi que des techniques de cytofluorométrie de flux ou d’affinités ou pour trier les cellules selon ce marquage [12].
Le repérage de la desmine, protéine présente précocement dans la différenciation de la cellule musculaire, a été réalisé par l’utilisation d’un anticorps primaire antidesmine (clone D33-DAKO M0760) dilué au 1/ 100 comparé à un contrôle isotypique (IgG1 non couplé Becton Dickinson 349040 dilution 1/25). Cette technique
nécessite la fixation et la la perméabilisation des cellules pour un marquage intracellulaire avec étapes d’amplification.
Viabilité cellulaire
La viabilité cellulaire a été évaluée par quantification du nombre de cellules capables d’exclure l’iodure de propidium. Mise en contact avec l’iodure de propidium, agent intercalant de l’ADN, les cellules viables ne sont pas marquées après deux minutes d’incubation. Les cellules restantes marquées sont considérées comme non viables.
Le tri est ensuite effectué par cytofluorométrie.
Longueur des télomères
La mesure de la longueur des télomères reflète les capacités de réplications cellulaires [13]. La taille des télomères a été quantifiée chez trois patients et deux contrôles selon les méthodes décrites par ailleurs [14, 15].
Différenciation in vitro
L’évaluation des capacités des cellules obtenues à former des myotubes plurinucléés a été effectuée dans des conditions de culture de différenciation. Les cellules ensemencées en boîtes (25cm2 Falcon) et en puits (2,1 cm2 Falcon) à la densité de 20 × 104 cellules par cm2 au milieu de prolifération à partir de cellules ayant subi au moins cinq passages c’est-à-dire environ vingt divisions. Ce stade de prolifération correspondant au rendement maximum et serait celui d’utilisation des cellules dans un protocole futur. La différenciation a ensuite été observée à J0, J3, J6, J9, J12 par photographies au microscope à contraste de phase pour les boîtes et en même temps dans les plaques multi-puits après fixation préalable au méthanol. Sur ces cellules ont été également réalisés par techniques d’immunofluorescence des marquages de desmine, CD56, chaînes lourdes de myosine néonatales rapides et lentes et la comparaison qualitative et semi quantitative de ces marquages avec ceux réalisés sur des cellules témoins a été analysée à partir de photos numériques.
Différenciation in vivo
L’analyse des capacités fonctionnelles in vivo des cellules obtenues a été analysée par implantation dans des muscles de souris Rag2/γc-/- (Rag2) lignées présentant deux mutations l’une dans le gène d’activation de la recombinase2 et l’autre dans le gène du récepteur commun des cytokines. De ce fait ces animaux ne développent pas de système immunitaire de type T ni de type B et ne réalisent pas de cytotoxicité dépendante de l’antigène [16]. Ce modèle animal a été choisi afin de reproduire les conditions identiques rencontrées dans une situation d’auto greffe.
Deux protocoles ont été utilisés pour injecter le muscle tibialis anterior chez la souris.
protocole 1 — injection d’environ 5×106 cellules diluées dans 20 ml, en 8 à 10 sites, par capillaire de verre fin sous contrôle de la vue après incision de la peau selon le protocole décrit par Kinoshita et col [17].
Protocole 2 — injection préalable de notexine (100 ng dilués dans 10 ml) dans le muscle en deux sites. La notexine est une toxine de venin de serpent induisant une dégénérescence-régénérescence complète du muscle reproduisant les conditions observées dans la dégénérescence de la dystrophie musculaire humaine.
Dans les deux cas la qualité de l’implantation a été jugée à quatre semaines par analyse histologique du tissu avec marquages par anticorps spécifiques (anti dystrophine, Novocastra NCL-Dys3) dirigés contre la partie C-terminale de la dystrophine humaine et révélés par anticorps secondaires fluorescents.
RÉSULTATS
Les résultats des analyses de sélection des patients ne sont pas montrés ici. Nous nous sommes assurés dans tous les cas que le tissu prélevé ne présentait aucun caractère lésionnel ni macroscopique à partir de l’observation d’IRM musculaire montrant l’absence de toute infiltration fibro-adipeuse de la zone prélevée, ni microscopique, l’étude anatomopathologique d’une fraction de l’échantillon biopsié ne montrant aucune altération dystrophique ni d’infiltrat inflammatoire.
Réalisation des cultures et rendement
Dans toutes les cultures, après une phase de latence initiale, une croissance rapide a été observée. Le nombre d’expansion a volontairement été limité à cinq. En trois ou quatre semaines le nombre de cellules produites était pour l’ensemble des cultures de l’ordre du milliard. Les courbes de croissance sont présentées dans le graphique figure 1 qui compare les rendements cellulaires moyens entre patients DMFSH et contrôles lors des différents passages. Le nombre de cellules qui avait été jugé optimal pour une réimplantation ultérieure a été atteint pour chacun des sujets prélevés en trois ou quatre semaines à partir d’une biopsie de taille raisonnable représentant entre 0,91 et 1,15 g de poids sec.
Évaluation des rendements théoriques
Évalués pour des séries d’expansion successives avant le ralentissement fort de la croissance des cellules, ces rendements sont calculés arbitrairement après arrêt lorsque le pourcentage de cellules myogéniques, évalué sur le marquage CD56+ et confirmé par le marquage desmine positif, passe en dessous de 50 %. Les chiffres obtenus sont variables. Aucune corrélation n’a pu être faite entre ces variations, le statut génétique, l’état pathologique ou l’âge du patient.
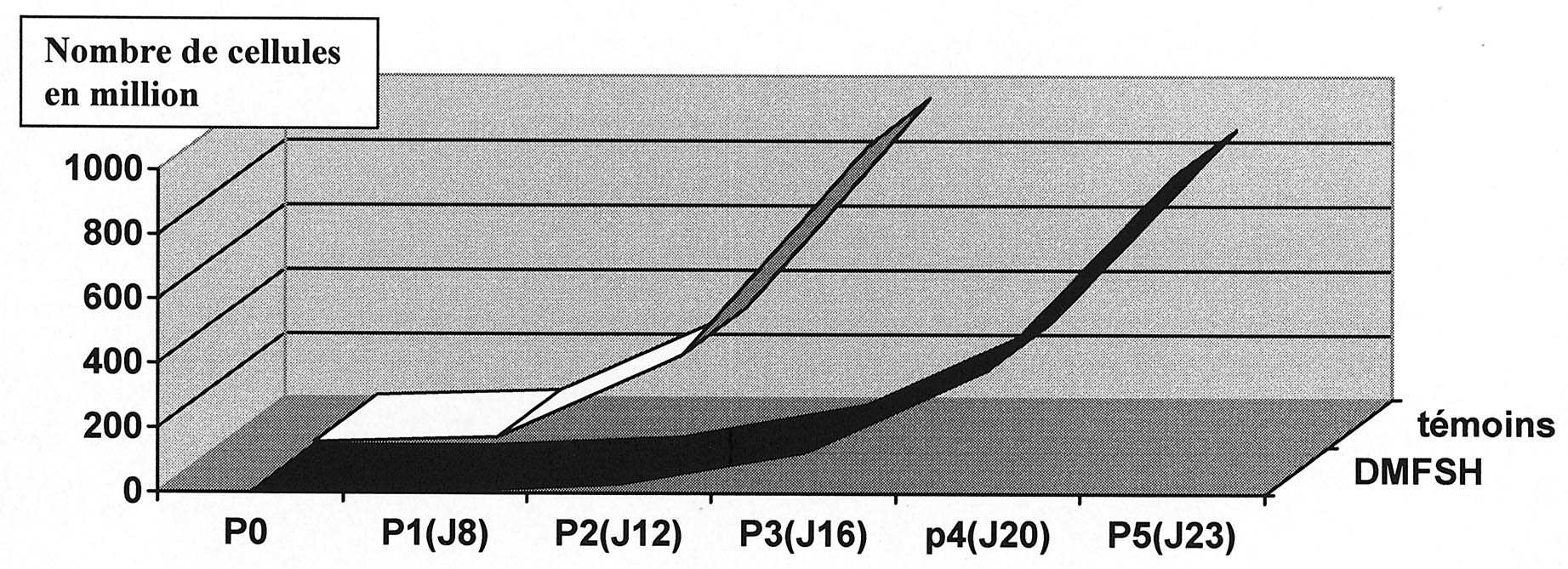
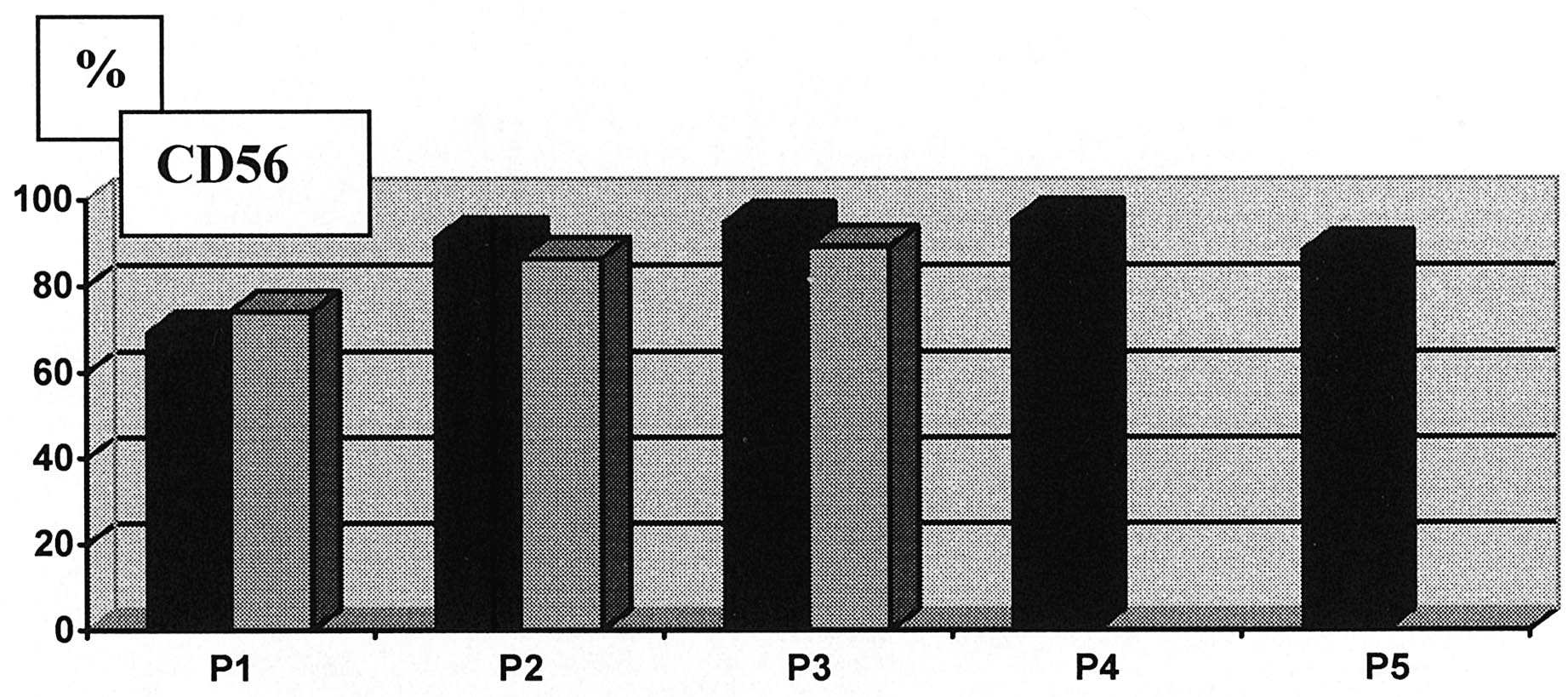
FIG. 1. — Cinétique de croissance des cellules (P = numéro de passage, J age en jour), en blanc :
cultures témoins, en noir : cultures patients DMFSH.
(Le nombre de cellules totales obtenues est similaire entre les témoins et les cultures de myoblastes de patients arrivant à un total d’environ 1000 millions de cellules. Le nombre optimal est obtenu après 3 passages pour les témoins, 5 passages pour les patients. Cette différence s’explique par la quantité de matériel disponible : 12 g en moyenne pour les témoins, 1 g en moyenne pour les patients).
FIG. 2. — Pourcentage de cellules exprimant le marquage CD56 avec l’évolution de l’expansion selon les passages. (En noir myoblastes à partir de patients MDFSH, en grisé myoblastes de sujets témoins. À partir du 2° passage de 80 à 9O % des cellules sont CD56 spécifiques des cellules myogéniques).
Caractérisation phénotypique
Issues des expansions, les cultures obtenues étaient très riches en cellules CD56+ contenant de 71 à 95 % de ce type cellulaire. L’expression CD56 et desmine augmentent parallèlement au cours des différents passages. Les histogrammes présentés dans la figure 2 montrent que, par comparaison avec les cellules témoins, les pourcentages sont similaires, plus variables au premier passage du fait du petit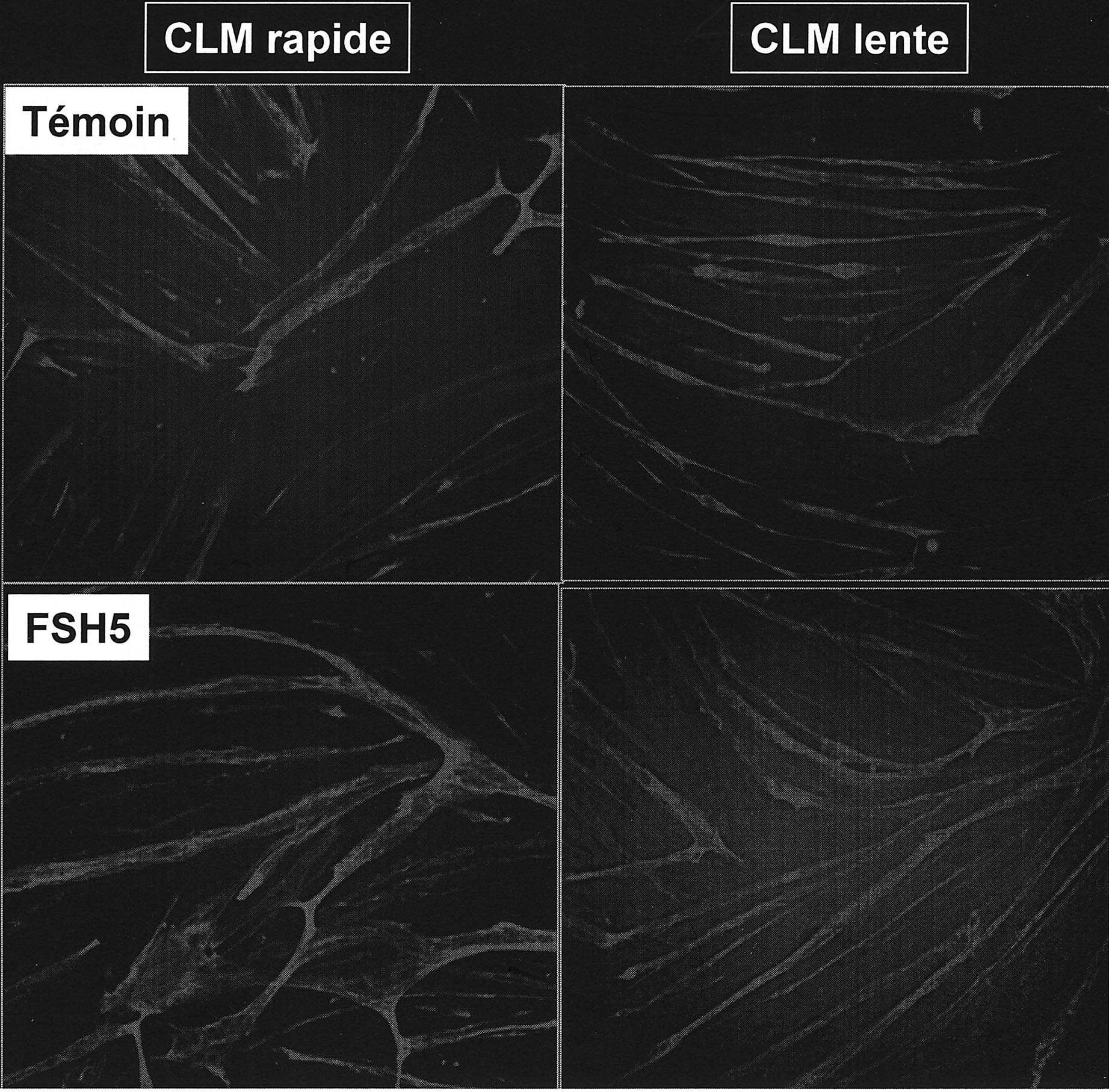
FIG. 3. — Différenciation en myotubes in vitro.
Photographies (contraste de phase, objectif × 20) des cultures témoins et patients DMFSH à un stade de différenciation avec confluence en myotubes après 3 jours en milieu de différenciation de cellules prélevées au stade 5° passage de prolifération. On n’observe aucune différence morphologique. Immunomarquage avec des AC spécifiques anti myosine lente (CML lente) et rapide (CML rapide). Ces protéines apparaissent normalement à ce stade de différenciation des myotubes humains. L’expression de ces protéines est identique dans les 2 préparations).
nombre de cellules obtenues puisque la technique utilisée requiert un important nombre de cellules et que de nombreuses cellules sont perdues aux étapes de fixation, perméabilisation et lavages successifs. D’une façon globale, le plus haut pourcentage est obtenu pour les deux marqueurs entre le 12 et 15ème jour pour l’ensemble des patients et ce pourcentage se maintient en plateau. À l’issue de la culture il est donc possible de préparer un nombre équivalent de cellules présentant une proportion identique de myoblastes CD56+ et desmine positif pour les cellules issues des patients DMFSH et pour les témoins.
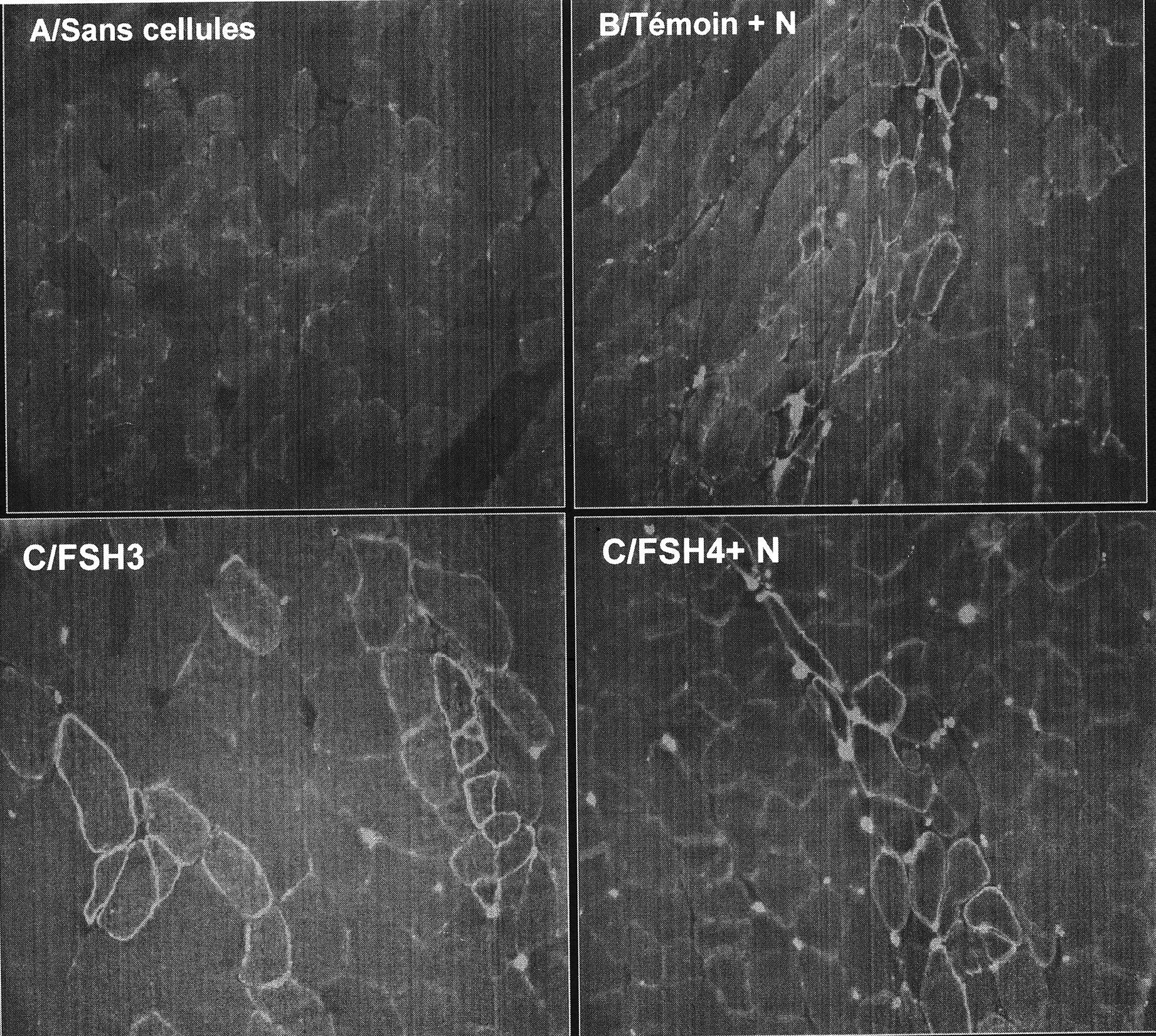
FIG. 4. — Différenciation in vivo chez la souris immunodéfficiente.
(Coupes transversales du muscle tibialis antérieur chez les souris RAG2 à 1 mois après implantation de 5×106 cellules au stade P5, immunomarquage avec anticorps anti-dystrophine humaine (protocoles injections et marquage dans section méthodes). A/ contrôle : injection du milieu sans cellule. B/ injection de cellules sujets témoins. C/ injection de cellules obtenues à partir du patient FSH3. D/ Injection de cellules obtenues à partir du patient FSH4. (+N = avec injection préalable de notexine). Rehaussement membranaire par fluorescence de la dystrophine humaine indiquant que ces cellules sont issues de différenciation des cellules humaines implantées chez les souris).
Taille des télomères
La taille des télomères après cinq à six passages en culture a été mesurée respectivement de 7,9 fi 0,1, 6,8 fi 0,3 et 7,9 fi 0,3 chez lez patients FSH1, FSH4 et FSH5.
Avec une moyenne 7,53 il n’y pas de différence significative avec les muscles témoins (6,9 fi 0,2 et 7,4 fi 0,3) ni avec des valeurs références de la littérature [15].
Différenciation in vitro
En condition de différenciation, les myoblastes issus de patients DMFSH se diffé- rencient et forment des myotubes dans un délai de 48 à 72 heures (figure 3). Ce temps de différenciation est identique à celui obtenu chez les contrôles. L’analyse qualitative réalisée sur les photos montre que l’expression par les myoblastes et les myotubes de desmine, de CD56, des isoformes des chaînes lourdes des myosines néonatales rapides et lentes est similaire dans les cultures différenciées de cellules des patients et des sujets contrôles Différenciation in vivo
L’expression de la dystrophine humaine a été analysée un mois après l’implantation dans les muscles de souris déficientes. Dans tous les cas l’injection de cellules humaines chez l’animal a permis l’expression de dystrophine humaine dans un nombre variable de fibres musculaires chez l’hôte. Des fibres hybrides et de nouvelles fibres ont été observées. Des injections ont été réalisées dans la patte contro-latérale chez chaque animal avec du milieu de culture seul et aucun marquage n’a été observé.
Nous avons utilisé des techniques d’observation qui ne permettent pas une quantification globale mais une analyse par champs successifs. Par champ microscopique, le nombre de cellules marquées est plus important dans le protocole notexine que dans le protocole sans notexine. Nous n’avons pas mis en évidence de différence lorsqu’il s’agissait de cellules contrôles et de cellules provenant de patients DMFSH.
La présence de groupements de fibres positives, de préférence en séries entre des fibres musculaires non transduites, suit le trajet de l’aiguille d’injection. On observe en effet plus de groupes de fibres positives que de fibres dispersées. Ces résultats sont montrés sur les photographies présentées dans la figure 4.
DISCUSSION
Malgré l’avancée des connaissances sur les mécanismes physiopathologiques des dystrophies musculaires il n’y a pas à ce jour de solution thérapeutique envisagée pour la DMFSH. Une technique orthopédique symptomatique de fixation de l’omoplate est utilisée de puis plusieurs années [18]. En raison de la présence fréquente d’une réaction macrophagique de type inflammatoire à l’examen anatomopathologique des biopsies musculaires de patients FSH, une corticothérapie a été proposée sans résultats [19]. Pour ses propriétés de renforcement des capacités musculaires la créatine a été également essayée mais sans succès [20]. Une très récente étude portant sur les effets conjugués du renforcement musculaire couplé à la prise d’albutérol ne conclut pas à recommander ce protocole [21]. La thérapie génique n’est pas envisageable ici puisque les données moléculaires restent fragmen-
taires. L’utilisation de cellules embryonnaires reste problématique dans les dystrophies musculaires, montrée non utilisable dans une étude très récente sur un modèle canin de la dystrophie musculaire de Duchenne [22] comme sur un modèle murin de sarcoglycanopathie [23].
C’est pourquoi, vu le caractère circonscrit de l’atteinte musculaire chez un individu donné, une solution thérapeutique possible dans la DMFSH serait une thérapie cellulaire par transfert autologue de myoblastes à partir d’un prélèvement tissulaire en zone phénotypiquement saine vers un territoire en dégénérescence, amyotrophique et déficitaire. Étant donné la situation autologue, les cellules transférées seront porteuses du trait génétique et il ne s’agira pas d’un traitement curatif mais d’un apport local de matériel cellulaire susceptible de renforcer ponctuellement les capacités régénératives dans un objectif thérapeutique symptomatique, possiblement limité dans le temps. Notre étude a ainsi été réalisée afin de savoir si les myoblastes porteurs du trait génétique DMFSH étaient susceptibles de régénération et de différenciation et pouvaient être utilisés dans les conditions thérapeutiques envisagées.
Sur des modèles animaux déficients, la transplantation de myoblastes permet un certain degré de régénération musculaire ainsi qu’une restauration d’expression protéique [24-27]. Chez l’homme, plus de 70 sujets atteints de dystrophie musculaire de Duchenne ont, au cours de ces dix dernières années, reçu des myoblastes issus de prélèvements musculaires squelettiques réalisés chez des donneurs compatibles.
Faits positifs, aucun effet secondaire en terme d’infection ou de carcinogenèse n’a été rapporté et la réimplantation a été localement bien tolérée. Par contre les résultats sont restés négatifs en ce qui concerne la réapparition d’un tissu fonctionnel jugé ici sur l’expression de la dystrophine, protéine manquante dans cette maladie [28-30]. Mais il faut noter d’une part la disparité des protocoles tant de préparation cellulaire que de réinjections ou d’évaluation et d’autre part l’utilisation d’immunosuppresseurs qui ont été mal tolérés et dont l’effet sur la prolifération cellulaire reste sans doute délétère. Il faut remarquer aussi que les objectifs thérapeutiques sont différents notre but n’étant pas de faire s’exprimer de novo une protéine absente suite à une modification génétique mais de renforcer les capacités régénératives d’un tissu musculaire placé dans un processus lent de dégénérescence génétiquement induite et dont les capacités de régénérescence deviennent progressivement in situ dépassées.
Dans la prévention de l’insuffisance cardiaque après infarctus du myocarde, l’objectif de renforcement semble avoir été atteint. Des études menées pour savoir s’il était possible d’augmenter la contractilité de la cicatrice de l’infarctus du myocarde ont montré que la transplantation de myoblastes adultes autologues dans la zone infarcie améliorait la fonction myocardique avec reprise d’activité métabolique de cette zone et persistance au long court des cellules injectées [31, 32].
Les difficultés rencontrées dans la thérapie cellulaire des dystrophies musculaires de type Duchenne semblent liées au mécanisme pathogénique de cette maladie. Plu-
sieurs études ont été conduites pour caractériser la croissance et l’évolution in vivo des cellules musculaires squelettiques à partir de biopsies musculaires prélevées chez des patients atteints. Comparé à des cellules témoins non dystrophiques, le temps de doublement des cultures est allongé, des phénotypes nouveaux apparaissent avec figure de sénescence, les télomères sont raccourcis, des caractéristiques de différenciation morphologique des myotubes sont altérées de même que certaines activités enzymatiques [33]. On explique ainsi que si le potentiel de prolifération cellulaire de régénérescence reste très élevé chez l’homme normal quel que soit l’âge, la multiplication compensatrice très élevée des myoblastes chez les sujets Duchenne provoque un épuisement rapide du cycle dégénérescence-régénérescence. Cependant, les myoblastes des patients DMD restent fonctionnellement capables après leur injection de participer à la fonction de tissu différencié et à l’expression des protéines associées à cet état [33]. Dans la dystrophie myotonique de Steinert, les myoblastes issus de patients atteints sont capables de faire apparaître, après réimplantation, un tissu musculaire myotonique [34] prouvant ainsi que lors de la réimplantation de myoblastes les caractéristiques fonctionnelles du tissu musculaire différencié d’origine sont conservés.
Dans la présente étude, les résultats obtenus à partir de cinq patients atteints DMFSH ont montré que la méthodologie mise en œuvre permet d’obtenir dans des délais raisonnables, une grande quantité de cellules, à partir d’une biopsie humaine de petite taille compatible avec la pratique clinique. Les cultures de myoblastes obtenues présentent un degré de pureté élevé (plus de 80 % de cellules CD56+ similaire aux cellules témoins). Ces cellules ne paraissent pas entrer plus rapidement en sénescence que les myoblastes non DMFSH comme l’indiquent les résultats des mesures de la longueur des télomères. Placé en milieu adapté, ces myoblastes ont fusionné et se sont différenciés en myotubes dans des délais comparables à celui des témoins et ces myotubes exprimaient des marqueurs de différenciation caractéristiques du stade post-myoblastique. Comme il n’existe pas de modèle animal de la DMFSH, nous avons choisi de tester les capacités régénératives in vivo dans le modèle murin immunodéficient Rag2 pour reproduire les conditions de réimplantation autologue. Quel que soit le protocole utilisé, des cellules musculaires de caractéristiques matures exprimant la dystrophine humaine sont apparues dans les fibres musculaires de l’hôte, qu’il s’agisse de fibres hybrides ou de fibres néoformées, ces dernières, situées dans l’épimysium, étaient cependant de petit diamètre, de formes irrégulières ou orientées dans le sens opposé à l’orientation générale du faisceau.
Cette étude, contrairement à celle publiée par Winokur et col [35], selon laquelle la myogénèse des myoblastes issus de patients DMFSH serait altérée, ne nous a pas fait observer ni de figure de nécrose ni de vacuolisation. Cette différence vient sans doute du fait que dans notre étude les myoblastes étaient prélevés dans une zone phénotypiquement indemne sans aucune manifestation clinique, radiologique ou histologique de dégénérescence, alors que l’origine n’est pas précisée dans l’étude de Winokur mais les faibles rendements cellulaires indiqués laissent à penser qu’il s’agit
de myoblastes provenant de tissu prélevé en zone dystrophique. Notre étude n’ayant pas été prévue dans un objectif physiopathologique, nous n’avons pas comparé les caractéristiques de myoblastes d’origine différente, mais elle démontre que pour le matériel et dans les conditions expérimentales utilisées il n’est pas observé de trouble de prolifération ni de différenciation des myoblastes porteurs du trait génétique marqueur de DMFSH. Aucune accumulation toxique n’est décrite dans ces cellules, il n’est donc pas vraisemblable que la culture accélère un processus délétère qui se manifesterait ultérieurement de manière tissulaire ou macroscopique. Dans la mesure où elles sont porteuses du trait génétique, on est en droit de se poser la question de leur devenir en tant qu’outil thérapeutique c’est-à-dire de leur persistance à moyen et long terme dans les bonnes conditions observées ici, mais il n’est pas possible de répondre à cette question en utilisant un modèle. Il n’existe pas à ce jour de modèle animal DMFSH. Utiliser un modèle murin d’une autre dystrophie musculaire ne serait pas informatif car il s’agirait de mécanismes physiopathologiques différents. Ainsi par exemple, le muscle du modèle mdx de la maladie de Duchenne présente une fragilité toute particulière à l’exercice excentrique tout en développant une fibrose modérée au contraire du modèle dy où les fibres restent non sensibles à ce type d’exercice alors qu’elles développent une fibrose très importante.
Utiliser un modèle murin de calpaïnopathie permettrait de tester une situation où l’atteinte reste restreinte à certains muscles [36] mais on sortirait d’une situation de greffe autologue. Aucun modèle de pathologie musculaire n’étant immunodéficient il n’est pas intéressant non plus d’utiliser sur des temps plus longs le modèle immunodeficient Rag car on introduirait un biais de sélection négative des cellules dystrophiques dans un environnement de cellules saines. De même, utiliser des techniques d’irradiation pour limiter les phénomènes de rejet sur des modèles dystrophiques ou des souris saines ne permettrait pas de prolonger l’observation de plus de quelques mois avant l’atrophie progressive induite par l’irradiation. Il apparaît donc que quelle que soit la méthode expérimentale envisagée les résultats en resteraient non informatifs et que seule une étude en situation réelle peut permettre d’avancer, tout en sachant que les cellules obtenues ici possèdent des caractéristiques de croissance identiques à celles utilisées dans l’étude de renforcement myocardique post ischémique et qu’il est donc vraisemblable que leur comportement à moyen terme soit similaire.
L’idée d’une thérapie cellulaire des dystrophies musculaires date déjà de plus d’une décennie [37] et après des résultats prometteurs sur les modèles animaux l’application à l’homme est restée décevante dans la maladie de Duchenne bien que l’amé- lioration des techniques de réimplantation semble permettre de nouveaux espoirs [38]. Il ne faut sans doute pas tourner la page trop vite et garder à l’esprit que cette maladie, si elle est le chef de file des dystrophies par déficit d’expression d’une protéine du complexe membranaire, reste très différente dans sa physiopathologie d’autres dystrophies comme la DMFSH où le mécanisme moléculaire causal semblerait mettre en jeu une régulation d’expression de protéines nucléaires. L’objectif de traitement n’est pas non plus similaire car nous n’envisageons pas une restaura-
tion des capacités « normales » du tissu musculaire mais introduisons un concept de biothérapie symptomatique. Notre étude bénéficie des avancées récentes de technologies de prolifération cellulaire. Dans les conditions fixées, les résultats obtenus in vitro dans le modèle de xenotransplantation immunodéficient utilisé sont une réelle avancée car permettent d’envisager un transfert vers une phase clinique de réimplantation humaine. D’un autre côté, les techniques récentes d’injections multipoints, validées chez le primate, permettent d’obtenir de biens meilleurs rendements de la réimplantation intra musculaire squelettique [39] que ceux obtenus jusqu’à présent mais reste à valider les études complémentaires entreprises pour tester d’autres conditions environnementales de réimplantation.
Remerciements
Ce travail a été financé par l’Association Française contre les Myopathies (AFM) et réalisé grâce à une promotion de la DRC du CHU de Nice. Les auteurs remercient Vincent Mouly et Gillian Butler-Browne (UMR CNRS 7000) pour la réalisation des mesures de longueur de télomères, et James Di Santo (Institut Pasteur) pour avoir mis à notre disposition les souris Rag.
BIBLIOGRAPHIE [1] PADBERG G. — Facio scapulo humeral disease. The Netherlands, Leiden University 1982 [2] TAWIL R. —Facioscapulohumeral Muscular Dystrophy. Curr. Neurol. Neurosci. Reports 2004, 4, 51-54.
[3] WIJMEGA C., HEWITT JE., SANDKUIJL A., CLARK LN., WRIGHT TJ., DAUWERSE HG. et al . —
Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy Nature Genet ., 1992, 2 , 26-30.
[4] LEMMERS RJ., VAN DER MAAREL S., VAN DEUKETOM JC., VAN DER WIELEN MJ., DEIDDA G., DAUWERSE HG. et al . — Inter and intrachromosomal subtelomeric rearrangements on 4q35 :
implications for facioscapulohumeral muscular dystrophy (FSHD) etiology an ddiagnosis.
Hum. Mol. Genet., 1998, 7 , 1207-1214.
[5] TAWIL R., FORRESTER J., GRIGGS RC., MENDELL JR., KISSEL J., MCDERMOTT MP. et al. —
Evidence for anticipation : an association of deletion size with severity in facioscapulohumeral muscular dystrophy. Ann. Neurol. , 1996, 39 , 744-748.
[6] GABELLINI D., GREEN MR., TUPLER R. — Inappropriate gene activation in FSHD : a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell., 2002, 110 , 339-348.
[7] TUPLER R., GABELLINI D. — Molecular basis of facioscapulohumeral dystrophy.
Cell. Mol. Life
Sci., 2004, 61 , 557-566.
[8] MAURO A. — Satellite cell of skeletal muscle fibres
J. Biophys. Biochem. Cytol ., 1961, 9 , 493-495.
[9] WRIGHT WE. — Myoblast senescence in muscular dystrophy.
Exp. Cell. Res., 1985, 157, 343-354.
[10] VILQUIN JT., MAROLLEAU JP., TREMBLAY JP., ROBERT I., TERNAUX B. — Procédé d’obtention de populations cellulaires d’origine musculaire et utilisations. Brevet (007304) déposé en France le 7 Juin 2000 et aux USA le 16 Octobre 2000 sous l’égide de l’AP-HP, l’INSERM et l’AFM.
[11] BELLES-ISLES M., ROY R., DANSEREAU G., GOULET M., ROY B., BOUCGARD JP., TREMBLAY JP.
— Rapid selection of donor myoblast clones for musclar dystrophy therapy using cell surface expression of NCAM. Eur. J. Histochem., 1993, 37 , 375-380.
[12] WEBSTER C., PAVLATH GK., PARKS DR., WALSH FS., BLAU HM. — Isolation of human myoblasts with the fluorescent- activated cell sorter. Exp. Cell Res ., 1988, 174 , 252-265.
[13] ALLSOPP RC., VAZIRI H., PATTERSON C., GOLDSTEIN S., YOUNGLAI EV., FUTCHER AB. et al. —
Telomere length predicts replicative capacity of human fibroblasts.
Proc. Natl. Acad. Sci. USA , 1992, 89 , 10114-10118.
[14] DECARY S., HAMIDA CB., MOULY V., BARBET JP., HETATI F., BUTLER-BROWNE GS. — Shorter telomeres in dystrophic muscle consistent with extensive regeneration in young children.
Neuromuscl. Disord ., 2000, 10 , 113-120.
[15] RENAULT V., THORNELL LE., ERIKSSON PO., BUTLER-BROWNE GS., MOULY V. — Regenerative potential of human skeletal muscle during aging. Aging Cell, 2002, 1 , 132-139.
[16] COOPER RN., IRINTCHEV A., DI SANTO JP., ZWEYER M., MORGAN JE., PARTRIDGE TA. et al. —
A new immunodefficient mouse model for human myoblast transplantation.
Hum. Gene Therapy , 2001, 12 , 823-831.
[17] KINOSHITA I., VILQUIN JT., GUERETTE B., ASSELIN I., ROY R., TREMBLAY JP. — Very efficient myoblat allotransplantation in mice under FK506 immunosuppression. Muscle Nerve , 1994, 17 , 1407-1415.
[18] LETOURNEL E., FARDEAU M., LYTLE JO., SERRAULT M., GOSSELIN RA. — Scapulothoracic arthrodesis for patients who have facioscapulohumeral muscular dystrophy. J. Bone Joint Surgery, 1990, 72 , 78-84.
[19] TAWIL R., MCDERMOTT MP., PANDYA S., KISSEL J., GRIGGS RC., and the FSHD-DY Group. — A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. Neurology, 1997, 48 , 46-49.
[20] WALTER MC., LOCHMULLER H., REILICH P.,KLOPSTOCK T., HUBERT R., HARTARD M. et al. —
Creatine monohydrate in muscular dystrophies : a double blind placebo controlled clinical study. Neurology, 2000, 54 , 1848-1850.
[21] VAN DEN KOOI EL., VOGELS OJ., VAN ASSELDONK RJ., LINDEMAN E., HENDR JC., WOHLGEMUT M. et al. — Strength training and albuterol in facioscapulohumeral muscular dystrophy.
Neurology, 2004, 63 , 702-708.
[22] DELL’AGNOLA C., WANG Z., STORB R., TAPSCOTT SJ., KUHR CS., HAUSCHKA SD. et al. —
Hematopoietic stem cell transplantation does not restore dystrophin expression in Duchenne Muscular Dystrophy dogs. Blood , 2004, 182 , 4311-4318.
[23] COSSU G. — Fusion of bone marrow derived stem cells with striated muscle may not be sufficient to activate gene expression. J. Clin. Invest., 2004, 114 , 1540-1543.
[24] PARTRIDGE TA., MORGAN JE., COULTON GR., HOFFMAN EP., KUNKEL LM. — Conversion of mdx myofibres from dystrophin-negative to dystrophine-positive by injection of normal myoblasts, Nature , 1989, 337 , 176-179.
[25] HUARD J., VERRAULT S., ROY R., TREMBLAY M., TREMBLAY JP. — High efficiency of muscle regeneration after human myoblats clone transplantation in SCID mice. J. Clin. Invest , 1994, 93 , 586-599.
[26] NEUMEYER AM., CROS D., MCKENNA-YASEK D., ZAWADSKA A., HOFFMAN EP., PEGORARO E. et al . — Pilot study of myoblast transfer in the treatment of Becker muscular dystrophy Neurology, 1998, 51 , 589-592.
[27] ITO H., VILQUIN JT., SKUK D., ROY B., GOULET M., LILLE S. et al. — Myoblast transplantation in non dystrophic dog.
Neuromuscul. Disord., 1998 , 8 , 95-110.
[28] GUSSONI E., PAVLATH GK., LANCTOT AM., SHARLA KR., MILLER RG. et al. — Normal dystrophin transcripts detected in Duchenne musculare dystrophy patients after myoblats transplantation. Nature , 1992, 356 , 435-438.
[29] KARPATI G., AJDUKOVIC D., ARNOLD D., GLEDHILL RB., GUTTMANN R., HOLLAND P. et al. —
Myoblast tranfer in Duchenne muscular dystrophy.
Ann. Neurol., 1993, 34 , 8-17.
[30] LAW PK., GOODWIN TG., FANG Q., QUINLEY T., VASTAGH G., HALL T. et al. — Human gene therapy with myoblast transfer.
Transplant. Proc., 1997, 29, 2234-2237.
[31] MENASCHÉ P., HAGEGE AA., SCORSIN M., POUZET B., DESNOS M., DUBOC D. et al. — First successful clinical myoblast transplantation for heart failure. The Lancet , 2001, 357 , 279-280.
[32] MENASCHÉ P., HAGEGE AA., VILQUIN JT., DESNOS M., ABERGEL E., POUZET B., et al. —
Autologous skeletal myoblast transplantation for sever post infarction left ventriular dysfunction. J. Am. Coll. Cardiol., 2003, 41 , 1078-1083.
[33] WESBSTER C., BLAU HM. — Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts : implications for cell and gene therapy. Somat. Cell Mol. Genet. , 16 , 557-565.
[34] SKUK D., FURLING D., BOUCHARD JP., GOULET M., ROY B., LACROIX Y., et al . — Transplantation of human myoblasts in SCID mice as a potential muscular model of myotonic dystrophy.
Neuropathol. Exp. Neurol., 1999, 58 , 921-931.
[35] WINOKUR ST., BARRETT K., MARTIN JH., FORRESTER JR., SIMON M., TAWIL R. et al. —
Facioscapulohumeral dystrophy (FSHD) myoblasts demontrate increased susceptibility to oxidative stress. Neuromusc. Disorders, 2003, 13, 322-333.
[36] RICHARD I., ROUDAUT C., MARCHAND S., BAGHDIGUIAN, HERASSE M., STOCKHOLM D. et al. —
Loss of calpain3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated κβα/nuclear factor kappaβ pathway perturbation in mice. J. Cell. Biol., 2000, 151 , 1583-1590.
[37] SKUK D. — Myoblast transplantation for inherited myopathie : a clinical approach.
Expert
Opin. Biol. Ther., 2004, 4 , 1871-1885.
[38] SKUK D., ROY B., GOULET A., CHAPDELAINE P., BOUCHARD JP., ROY R. et al . — Dystrophin expression of myofibers of Duchenne muscular dystrophy patients following intra muscular injections of normal myogenic cells. Mol. Ther., 2004, 9 , 475-482.
[39] SKUK D., GOULET M., ROY B., TREMBLAY JP. — Efficacy of myoblast transplantation in whole muscle of non-human primates following simple intramuscular injections : toward defining strategies applicable to humans Exp. Neurol., 2002, 175 , 112-126.
DISCUSSION
M. Philippe MONOD-BROCA
L’historique n’est certes pas l’essentiel de cette communication, mais puisque l’auteur a cité quelques noms, pourquoi n’a-t-il pas évoqué celui de Paul Broca qui, à propos de l’observation de pieds bots a été le premier — c’était en 1847 — à parler de myopathies.
Merci pour cette précision. Hommage a été rendu à Louis Landouzy et à Jules Déjerine, pour leur description princeps de la maladie, lors d’une séance à l’Académie nationale de médecine, il y a 120 ans.
M. Michel FARDEAU
Les premiers essais de transfert myoblastique dans les dystrophies de Duchenne se sont soldés par des échecs, et ceci justifie l’importance des études précliniques comme celles conduites par Claude Desnuelle et son équipe dans la dystrophie facio-scapulo-humérale.
Cependant, dans ce dernier cas, les cellules greffées de façon autologue sont porteuses de l’anomalie génétique. Il convient donc de se poser la question de la durée d’observation des greffes réalisées chez la souris pour apprécier le potentiel dystrophique des cellules injectées ?
C’est effectivement la question primordiale. Nous avons montré qu’il n’y a pas de processus de vieillissement induit par la maladie ou par la culture. Dans la mesure où il n’y a pas de modèle animal de la DMFSH à ce jour, nous avons utilisé un modèle de souris non dystrophique, immunodéficiente, où, à un mois après injection de myoblastes porteurs du trait génétique de la DMFSH, persistent des manifestations de leur participation au processus régénératif musculaire. L’utilisation d’un modèle animal autre ne serait pas informative puisque la cause génétique et le mécanisme physiopathologique y seraient différents et introduiraient des biais expérimentaux. Quoiqu’il en soit l’objectif est ici de renforcer les capacités régénératives d’un muscle qui a manifestement perdu ses capacités à contrebalancer spontanément la dégénérescence induite par la mutation génétique causale. Même si l’effet de ce renforcement reste limité dans le temps et qu’il est nécessaire de procéder à de nouvelles réimplantations après un temps de répit, dans l’état actuel des possibilités thérapeutiques des dystrophies musculaires, ce procédé thérapeutique pourrait être un intéressant progrès.
M. Jacques-Louis BINET
Quelle est la fréquence de cette dystrophie musculaire facio-scapulo-humérale en France ?
Où en êtes-vous dans les protocoles thérapeutiques chez l’homme ?
La DMFSH est la troisième myopathie par ordre de fréquence après la dystrophie musculaire de Duchenne et la dystrophie myotonique de Steinert, sa prévalence est de 1 sur 20 000. Nous sommes actuellement sur le point de débuter un essai de phase I/II dont l’objectif principal est de tester la tolérance à l’injection de gros volumes cellulaires par de multiples injections.
M. Yves GROSGOGEAT
A-t-on une idée sur l’évolution structurale de ces myoblastes injectés ? Est-on certain qu’ils puissent s’ordonnancer pour aboutir à une véritable contraction homogène et efficace ?
Un concept similaire est en cours d’évaluation dans le traitement de la cicatrice de l’infarctus du myocarde par Philippe Ménasché. Les évaluations préliminaires réalisées par son équipe ont montré que du tissu musculaire squelettique se reconstituait à partir des myoblastes autologues injectés au sein du tissu myocardique. Par ailleurs, J.P.
Tremblay à Québec m’a personnellement rapporté très récemment qu’un patient atteint de dystrophie de Becker, traité dans son centre par injections de myoblastes hétérologues dans l’éminence thénar avait eu un renforcement de ses capacités musculaires, mais ceci reste à être confirmé par une étude en cours sur un nombre statistiquement significatif de sujets.
* Centre de Référence pour les Maladies Neuromusculaires — CHU de Nice. Groupe Hospitalier l’Archet, BP 3079, 06202 Nice cedex 03 et INSERM U 638. ** Laboratoire de Thérapie Cellulaire — Hôpital Saint Louis, 1 Avenue Claude Vellefaux, 75475 Paris cedex 10. *** INSERM U 582, Pathologies et Thérapies du Muscle Strié, Institut de Myologie, Hôpital Pitié Salpetrière, 47 Boulevard de l’Hopital, 75651 Paris cedex 13. Tirés-à-part : Professeur Claude DESNUELLE, adresse ci-dessus. Article reçu le 19 octobre 2004, accepté le 7 février 2005.
Bull. Acad. Natle Méd., 2005, 189, no 4, 697-714, séance du 19 avril 2005