
Résumé
La fonction biliaire est une fonction hépatospécifique, résultat d’un transport vectoriel de substrats endogènes et exogènes à travers trois compartiments en série : espace vasculaire, espace cellulaire (hépatocytaire) et espace biliaire. La fonction biliaire est vitale pour l’organisme. Elle assure en effet, l’homéostasie lipidique, en particulier l’homéostasie du cholestérol, l’élimination des endo- et xénobiotiques potentiellement toxiques, produits de dégradation de l’hémoglobine sous forme de bilirubine, produits de dégradation des bacté- ries (endotoxines), et l’élimination de nombreux médiateurs de l’inflammation. La bile est élaborée initialement dans les cellules parenchymateuses hépatiques (hépatocytes), puis modifiée par des mécanismes de sécrétion et de réabsorption des cholangiocytes (cellules épithéliales biliaires). Elle conditionne la digestion et l’absorption intestinales. Le principal déterminant de la formation de la bile est un processus de filtration osmotique dû au transport actif des acides biliaires et de solutés osmotiquement actifs. Les transporteurs membranaires assurant la formation de la bile sont maintenant en grande partie identifiés. L’expression de ces transporteurs membranaires est régulée à la fois par des mécanismes transcriptionnels et post-traductionnels. La régulation transcriptionnelle est sous le contrôle de récepteurs nucléaires activés par des ligands dont les principaux sont les acides biliaires, stéroïdes synthétisés dans l’hépatocyte à partir du cholestérol. Les maladies cholestatiques constituent un groupe de maladies d’origine génétique ou résultant de l’interaction entre des facteurs génétiques et de l’environnement. Les maladies génétiques monogéniques démembrées au cours des dernières années illustrent le rôle crucial des transporteurs membranaires dans la fonction biliaire. Les maladies biliaires cholestatiques ont en commun de progresser vers la cirrhose pouvant justifier la transplantation hépatique. Au cours de ces maladies, les acides biliaires et les médiateurs de l’inflammation modulent l’expression des gènes contrôlant les transporteurs biliaires et certains récepteurs nucléaires. Cette régulation peut être considérée comme une réponse adaptative et pouvant contribuer à la variabilité phénotypique de ces pathologies. Le traitement de ces maladies repose actuellement sur l’administration de l’acide ursodésoxycholique et sur la transplantation hépatique en cas d’échec de ce traitement médical. Les progrès récents devraient aboutir à la mise au point de nouveaux médicaments ciblés sur les anomalies moléculaires caractéristiques de ces entités pathologiques.
Summary
Biliary function is a vital function of the liver which results from the vectorial transport of endogenous and exogenous substrates through three compartments sequentially : the vascular space, the cellular space and the biliary space. The biliary function is responsible for the homeostasis of lipid metabolism in particular of cholesterol metabolism, the elimination of toxic endo— and xenobiotics such as (bilirubin, lipid bacteria products (endotoxin)) and several inflammatory mediators. Bile elaborated in canaliculi, is modified by cholangiocytes through secretion and absorption. Bile is essential for the intestinal digestion and absorption of nutriments. The main determinant of bile formation is an osmotic filtration process resulting from active transport of bile acids and other osmotic solutes (glutathion). Most of the membrane transporters ensuring bile formation have now been identified. The expression of these membrane transporters is regulated through transcriptional and posttraductional mechanisms. Transcriptional regulation is under the control of nuclear receptors activated by ligands such as bile acids, which act as endogenous steroids synthesized from cholesterol in hepatocytes. Cholestatic liver diseases comprise genetic diseases resulting from the complex interaction between genetic and environmental factors. Monogenic cholestatic diseases recently identified illustrate the key role of membrane transporters in biliary function. Bile acids and inflammatory mediators are potent modulators transporters and nuclear receptor genes and thus trigger an adaptative response to cholestasis. The extent of this adaptative response could explain the compelling phenotypic variability of cholestatic diseases in childhood and adults. The firstline medical treatment is currently ursodeoxycholic acid and in case of failure of this medical treatment, liver transplantation is required. Recent progress in the molecular pathogenesis of bile formation and cholestatic liver diseases is expected to provide the design of drugs targeted to the molecular abnormalities typical of cholestatic diseases.
INTRODUCTION
La fonction biliaire est une fonction hépatospécifique dont le rôle est d’assurer l’élimination des xénobiotiques et de certains endobiotiques potentiellement toxiques comme la bilirubine, le cholestérol et leurs produits de dégradation les acides
biliaires primaires ou secondaires. Cette fonction est assurée par le transport vectoriel de substrats à travers trois compartiments disposés en série : l’espace vasculaire, l’espace hépatocytaire et l’espace biliaire. L’espace vasculaire est constitué par les sinusoïdes dont le volume total est équivalent au volume hépatocytaire.
Les hépatocytes sont des cellules polarisées comprenant une membrane basolatérale en contact avec le sang sinusoïdal et un pôle luminal délimitant un espace clos de 2 à 3 µ, le canalicule biliaire. Les canalicules biliaires ont une longueur totale d’environ 2 kms et rejoignent les canaux biliaires interlobulaires, puis septaux et segmentaires, bordés par les cellules épithéliales biliaires ou cholangiocytes. La bile est élaborée initialement par les hépatocytes dans le canalicule biliaire. Elle est ensuite modifiée par les cholangiocytes par l’intermédiaire de mécanismes de de sécrétion et de réabsorption, au cours de son passage dans les canaux biliaires. Le principal déterminant de la formation de la bile est un processus de filtration osmotique. Les solutés actifs d’un point de vue osmotique sont transportés au pôle canaliculaire des hépatocytes par des mécanismes de transport requérant de l’énergie. On distingue classiquement deux fractions dans la sécrétion biliaire, la fraction dépendante des acides biliaires et la fraction indépendante. Cette dernière est assurée par le transport du glutathion dans les canalicules, du chlore, du bicarbonate et de l’eau dans les canaux biliaires.
LES TRANSPORTS HÉPATOBILIAIRES
La Figure 1 et le Tableau 1 listent les principaux transporteurs membranaires qui déterminent la fonction biliaire. Ils ont été clonés à partir du foie humain et du foie de rongeur. Plusieurs revues récentes font le point sur la fonction et la régulation de ces protéines membranaires de transport [1-3]. En bref, ces transporteurs sont pour certains d’entre-eux des membres de la famille des « Solute Carriers » (SLCs) dont le rôle est d’assurer le transport vectoriel des acides biliaires et des solutés organiques du sang portal vers l’hépatocyte. Les substrats de ces solutes carriers sont non seulement des endo- ou xénobiotiques mais également des médiateurs de l’inflammation (cytokines, leucotriènes…). Ces transporteurs comprennent les membres de la famille des OATP (organic anion transporter (SLC21) et NTCP (SLC10A1). Ils assurent la captation hépatocytaire des acides biliaires, de la bilirubine et de la majorité des xénobiotiques. L’exportation de l’hépatocyte vers l’espace canaliculaire est assurée par plusieurs transporteurs de la membrane canaliculaire appartenant à la superfamille ABC (ATP Binding Cassette). Ces transporteurs requièrent de l’énergie fournie par l’ATP. Les principaux transporteurs ABC sont MDR1 (ABCB1), BSEP (ABCB11) principal transporteur des acides biliaires, MRP2 (ABCC2) responsable du transport de la bilirubine conjuguée et du glutathion (c’est le principal déterminant de la fraction indépendante de la bile), MDR3 (ABCB4) responsable du transport des phospholipides. Certains transporteurs des acides biliaires et des solutés organiques sont également présents dans les cholangiocytes en particulier, ASBT (Apical Bile Salt Transporter) (SLC10A2). Situé sur la mem-
TABLEAU 1. — Nomenclature, localisation et fonctions des principaux transporteurs hépatocytaires et cholangiocytaires et des facteurs de transcription impliqués dans l’homéostasie biliaire.
brane luminale, sa fonction serait de réabsorber les acides biliaires de la bile en amont d’un obstacle sur les voies biliaires. Ce transporteur a été initialement identifié à la membrane apicale des cellules épithéliales de l’intestin grêle terminal où se fait la réabsorption active des acides biliaires. MDR1 (ABCB1) est également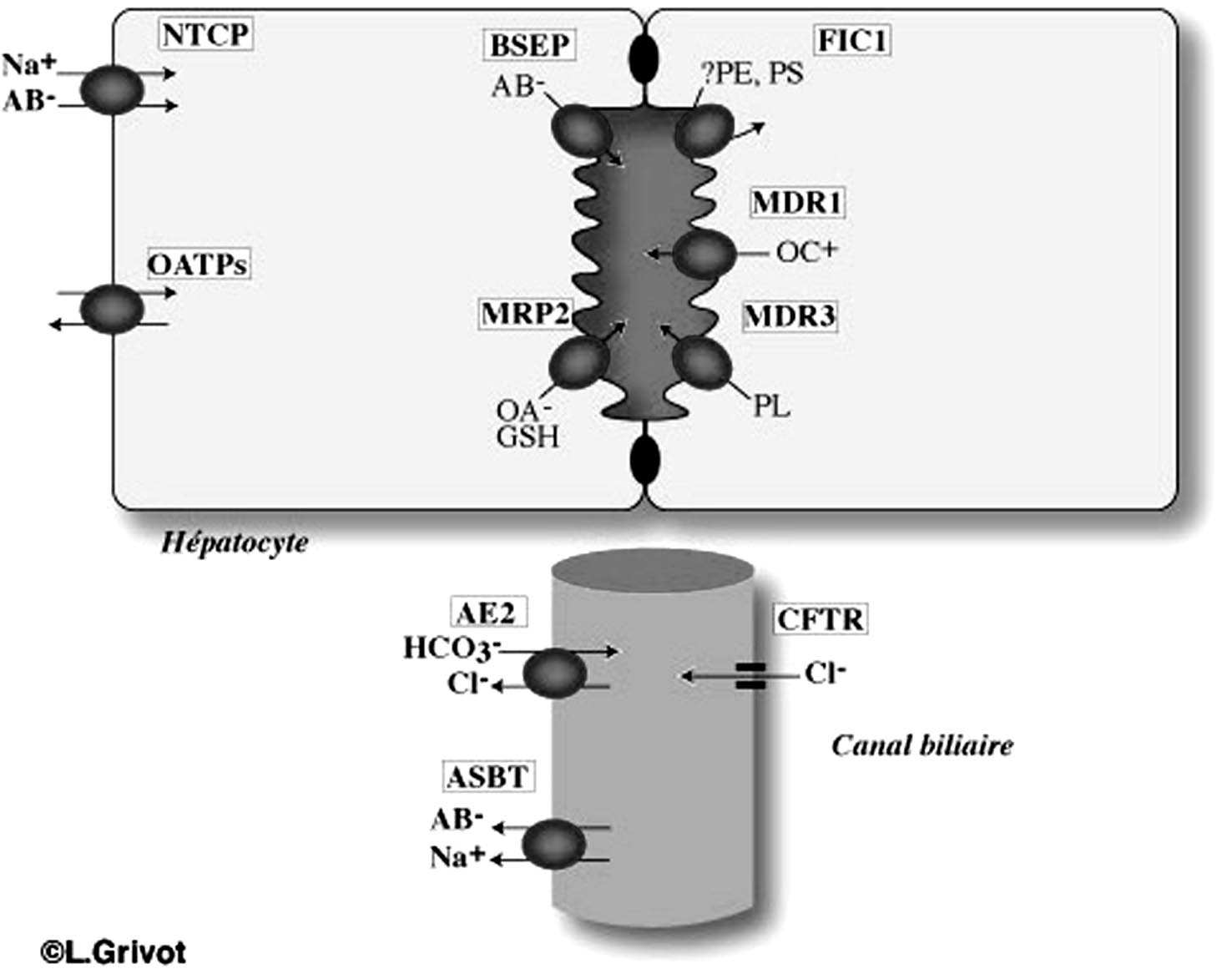
FIGURE 1. — Localisation des principaux transporteurs hépatobiliaires.
présent sur la membrane luminale des cholangiocytes et des cellules épithéliales de l’intestin grêle. Deux transporteurs CFTR (ABCC7) et AE2 (SLC4A2) situés sur la membrane luminale des cholangiocytes ont pour fonction d’assurer la sécrétion biliaire de bicarbonate.
Toutes ces protéines exprimées de façon constitutive par l’hépatocyte ou les cholangiocytes subissent une régulation transcriptionnelle et post-traductionnelle. La régulation transcriptionnelle est sous le contrôle de récepteurs nucléaires dont les ligands sont les acides biliaires, les oxystérols, la vitamine A et ses dérivés, les xénobiotiques.
LES RÉCEPTEURS NUCLÉAIRES
Les récepteurs nucléaires constituent une superfamille de facteurs de transcription activés par des ligands. Ils ont la propriété de réguler l’expression d’un grand nombre de gènes impliqués dans le développement, la physiologie et la toxicologie (4,8). Ces protéines contiennent une partie centrale hautement conservée faite
d’environ 70 acides aminés qui constituent le motif de liaison à l’ADN, disposés sous forme de 2 modules en doigt de zinc, critère qui définit cette superfamille de récepteurs. La partie C terminale comprend un domaine de liaison aux ligands moins conservé, responsable de la liaison aux hormones ou aux molécules hydrophobes, de la capacité de dimérisation et d’activation dépendante du ligand. A quelques exceptions près, ces récepteurs résident dans le noyau et se trouvent liés à des séquences d’ADN spécifique situées dans les régions flanquant 5′des gènes régulés par des récepteurs. En l’absence de ligand, ces récepteurs sont associés à des corépresseurs qui maintiennent le gène dans un état « off ». Une fois la liaison réalisée avec le ligand lipophile ou l’hormone, ces récepteurs nucléaires subissent des modifications conformationnelles avec relargage du corépresseur, liaison à une protéine coactivatrice qui permet l’interaction avec la machine transcriptionnelle du gène cible.
Les principaux récepteurs nucléaires impliqués dans l’homéostasie des acides biliaires est l’élimination des endo et xénobiotiques, sont le farnésoïd X réceptor (FXR), le Pregnand X Receptor (PXR), dont l’homologue humain est Steroid et Xenobiotic Receptor (SXR), Constitutive Androstane Receptor (CAR) et le Liver XR (LXR Êα et β).
INTERACTION RÉCEPTEURS NUCLÉAIRES-GÈNES DES TRANSPORTEURS RÉGULATION PAR LES RÉCEPTEURS NUCLÉAIRES
Les principaux ligands de FXR sont les acides biliaires hydrophobes, en particulier l’acide biliaire primaire de l’homme, l’acide chénodésoxycholique. La Figure 2 schématise le rôle de FXR dans l’homéostasie des acides biliaires. La liaison de FXR à son ligand, l’acide biliaire, entraîne une diminution de l’expression de NTCP ainsi que de CYP7A1 et de CYP8B1 respectivement la 7-alpha et 12-alphahydroxylase responsable de la synthèse des acides biliaires à partir du cholestérol dans l’hépatocyte. La liaison de l’acide biliaire à FXR et sa fixation sur les gènes cibles entraînent également une augmentation de la transcription et de l’expression de BSEP. Ainsi, FXR est le régulateur de la concentration intrahépatocytaire des acides biliaires. La fonction de FXR a, par ailleurs, été précisée à l’aide de souris knock-out pour ce facteur de transcription. Ces souris ont une accumulation d’acides biliaires dans le foie et dans le sang. Cette accumulation s’accentue avec l’âge et entraîne progressivement une atteinte hépatique qui devient sévère si les souris reçoivent un régime enrichi en acide cholique [9, 10].
SXR/PXR ont pour principaux ligands, certains xénobiotiques en particulier la rifampicine et les acides biliaires hydrophobes comme le lithocholate. Les principaux gènes cibles de PXR sont CYP3A4 et MRP2. Le CYP3A4 est responsable de l’hydroxylation et donc de l’élimination des acides biliaires hydrophobes. Les souris knock-out pour PXR sont incapables de métaboliser un grand nombre de médica-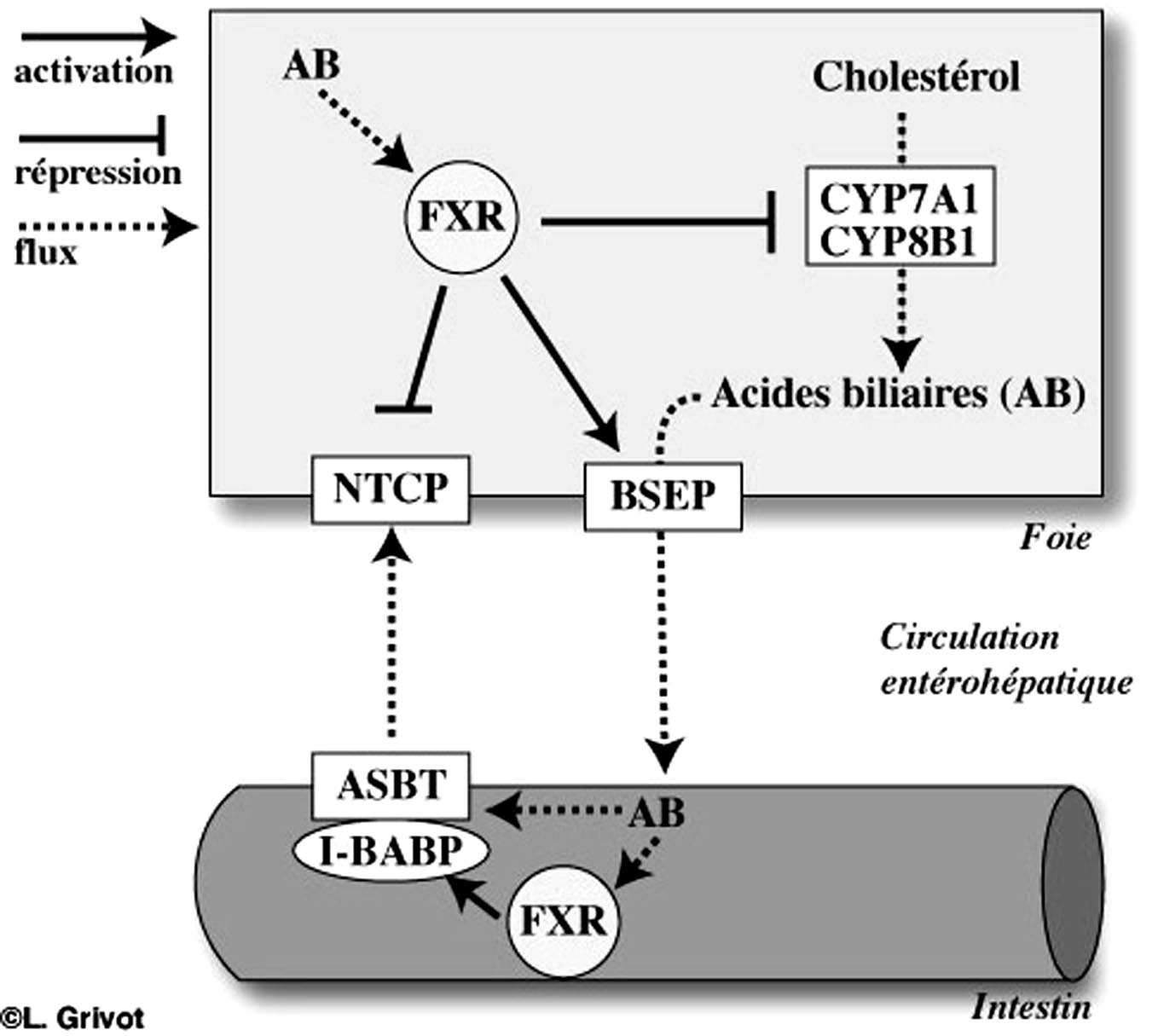
FIGURE 2. — Régulation de l’homéostasie biliaire par le récepteur nucléaire FXR.
ments et développent une atteinte hépatique sévère du fait d’un défaut de détoxification de l’acide lithocholique. PXR comme FXR a donc un rôle physiologie protecteur contre l’excès d’acides biliaires dans les cellules hépatiques [11-17].
CAR est un récepteur aux xénobiotiques dont les principales fonctions sont de réguler le métabolisme de la bilirubine et des médicaments. Ce facteur de transcription est actif de façon constitutive. Bien qu’activé par le phénobarbital, ce médicament n’est pas un ligand de CAR mais permet sa translocation du cytoplasme vers le noyau. Les principaux gènes cibles hépatocytaires de CAR sont les cytochromes 2B, 2C, 3A et 4A. CAR induit par ailleurs l’expression de l’ensemble des protéines impliquées dans la clairance la bilirubine. CAR est activé par les taux élevés de bilirubine intrahépatocytaire. Autrement dit, la bilirubine induit sa propre clairance. L’induction du cytochrome P450 par CAR est en principe une réponse protectrice vis-à-vis de certains xénobiotiques. Cependant, dans certains cas, l’activation de CAR aboutit comme cela est attendu à une production de métabolites
toxiques. C’est ainsi que l’utilisation d’agonistes inverses de CAR permet d’inhiber la toxicité induite par l’acétaminofène [17].
LXR constitue une classe de récepteurs nucléaires activés par les oxystérols. LXR alpha et bêta régule l’expression des gènes impliqués dans l’homéostasie lipidique.
LXR est exprimé non seulement dans le foie mais également dans d’autres tissus en particulier les macrophages, où les oxystérols sont présents du fait d’une internalisation des LDL oxydés ou générés in situ à partir de modifications du cholestérol.
Les gènes hépatiques cibles de LXR sont CYP7A1, ABCG5, ABCG8 (gènes impliqués dans le transport du cholestérol de l’hépatocyte vers le canalicule et impliqués dans la régulation l’absorption digestive du cholestérol et de l’efflux du cholestérol des macrophages). Au cours de la cirrhose biliaire primitive et des maladies inflammatoires des voies biliaires, les perturbations du métabolisme lipidique affectent environ 30 à 40 % de l’ensemble des malades. Dans le contexte de ces maladies inflammatoires, il est intéressant de noter que les ligands de LXR ont aussi pour effet d’inhiber l’expression des médiateurs de l’inflammation comme la NO synthase, la cyclooxygénase 2, et l’interleukine 6 dont l’expression est augmentée dans les maladies inflammatoires biliaires. Les souris LXR knock-out (LXR alpha -et LXR bêta-) ont été l’objet d’études portant sur le métabolisme du cholestérol et des acides biliaires. Le foie de ces souris accumule du cholestérol et peut développer une cirrhose. Cependant, l’étude fine de la fonction hépatobiliaire chez ces animaux est mal connue [18-24].
PHYSIOPATHOLOGIE ET ÉTIOLOGIE DES MALADIES BILIAIRES INFLAMMATOIRES
Dans l’état actuel de nos connaissances, ces maladies sont associées à des défauts de la sécrétion canaliculaire hépatocytaire ou à des anomalies des fonctions des cellules bordant les canaux biliaires, les cholangiocytes. Les maladies cholestatiques d’origine hépatocytaire résultent de mutations génétiques ou d’altération secondaire à des médicaments, à des toxiques ou à des infections virales. Les cholangiopathies sont représentées chez l’enfant principalement par la mucoviscidose, le syndrome d’Alagille, et les cholestases familiales progressives alors que les cholangiopathies de l’adulte sont principalement représentées par la cirrhose biliaire primitive et les cholangites sclérosantes, un ensemble de maladies inflammatoires associées très fréquemment à des entérocolites inflammatoires. Quelle que soit l’origine de la maladie, génétique ou acquise, ces maladies s’accompagnent de modifications de l’expression de la fonction des protéines régulant la fonction biliaire et l’homéostasie des acides biliaires et des lipides.
Mutations génétiques responsables des cholestases familiales progressives
Les mutations de plusieurs transporteurs ABC sont considérés comme responsables des cholestases familiales progressives (PFIC) chez l’enfant [25, 26]. Ainsi les
mutations d’un gène codant pour la protéine FIC1 présente dans la membrane canaliculaire et dans les cholangiocytes mais aussi dans le pancréas et l’intestin, est responsable de la maladie de Byler. Ce gène et la protéine n’ont pas été listés dans le texte plus haut car le rôle de la protéine FIC1 est mal connue. La cholestase familiale progressive de type 2 est également une maladie cholestatique sévère, conduisant à l’insuffisance hépatique. Des mutations de la protéine BSEP sont responsables du phénotype clinique de ces patients. Environ 30 mutations différentes ont été décrites.
La cholestase familiale de type 3 est secondaire à des mutations du gène MDR3. La protéine est absente ou faiblement exprimée dans le foie de ces patients. Au cours de cette maladie cholestatique, il existe une inflammation et une altération importantes des petites voies biliaires comme au cours de la cirrhose biliaire primitive. La bile de ces patients est caractérisée par une concentration très basse en phospholipides.
L’étude de ce gène nous a permis également de montrer qu’il était responsable d’une maladie de l’adulte jeune associant une microlithiase biliaire et une cholangiopathie parfois responsable d’atteinte hépatique sévère [27]. Les mutations de CFTR sont responsables d’un défaut de sécrétion de chlore par les cholangiocytes avec pour résultat une cholestase et une inflammation des voies biliaires touchant environ 10 à 20 % des patients ayant une mucoviscidose. Nous avons rapporté récemment le rôle aggravant potentiel des mutations de CFTR dans certaines formes de maladies inflammatoires des voies biliaires de l’adulte [28].
Ces exemples montrent que des altérations importantes des gènes codant les protéines régulant la fonction biliaire sont responsables de maladies sévères chez l’enfant, plus rarement chez l’adulte jeune suggérant que des altérations génétiques ou des polymorphismes pourraient expliquer l’expression phénotypique des maladies biliaires inflammatoires de l’adulte et en particulier de la cirrhose biliaire primitive.
Régulation et adaptation des transporteurs hépatobiliaires dans les modèles expérimentaux cholestases expérimentales et acquises chez l’homme
Il existe de plus au cours des maladies cholestatiques, une modification d’expression des transporteurs biliaires, qui peut être considérée comme un mécanisme de protection. [29-32].
S’agissant des transporteurs basolatéraux, dans toutes les formes de cholestase, il est observé une diminution de l’expression de NTCP aboutissant ainsi à une diminution de la captation des acides biliaires.
S’agissant des transporteurs canaliculaires, il a été montré que MDR1, le transporteur des cations organiques (colchicine, ciclosporine, doxarubicine, tamoxifène, tacrolimus etc..) est up-régulé au cours des maladies cholestatiques. L’expression hépatique de MDR3 dans les modèles expérimentaux est peu modifiée. L’expression de BSEP et généralement de MRP2 au pôle canaliculaire est longtemps conservée.
Autrement dit, l’adaptation des transporteurs a pour but de préserver l’hépatocyte
de l’accumulation intrahépatocytaire de toxiques qu’ils soient endogènes ou exogè- nes.
S’agissant des transporteurs intestinaux, des études préliminaires indiquent que MRP2 et MDR1 sont diminués dans l’intestin après cholestase par ligature section chez le rat. Les modifications de ASBT responsable de la réabsorption des acides biliaires sont controversées, mais les acides biliaires augmentent l’expression d’IBAP, protéine de liaison des acides biliaires à l’intérieur de l’entérocyte.
Le mécanisme de la régulation adaptative des transporteurs biliaires constitue une thématique émergente dont l’intérêt est évident puisque la connaissance de ces mécanismes ouvre la voie à des thérapeutiques médicales adaptées à chaque situation. L’état de nos connaissances peut être résumé de la manière suivante : au cours de la ligature section du cholédoque qui entraîne une cholestase, les transcrits de FXR et de RXR sont diminués et sont considérés comme responsables de la diminution de NTCP. Les agonistes de la protéine FXR augmentent l’expression de BSEP, MRP2 et MDR3. Les modifications des transcrits de PXR au cours des maladies cholestatiques ainsi que ceux de LXR ne sont pas connus. L’acide ursodé- soxycholique et la rifampicine, des ligands de SXR ont un effet bénéfique dans les maladies cholestatiques suggérant que ce facteur de transcription ou ses modifications ont un rôle clé dans l’expression clinique de ces maladies.
Les cholestases sont en outre associées à des modifications importantes du cytosquelette des hépatocytes : désorganisation des microtubules, augmentation des filaments intermédiaires et accumulation des microfilaments d’actine dans le domaine péricanaliculaire [33 ]. Ces modifications du cytosquelette expliquent la perte de la structure normale du canalicule biliaire et de ses microvillosités [34]. Le ciblage des composants membranaires, la transcytose et l’exocytose canaliculaire sont également perturbées expliquant la rétention des transporteurs canaliculaires à la surface basolatérale des hépatocytes. Ces vésicules contiennent en effet les protéines de transport synthétisées dans le noyau et vectorisées au canalicule. L’accumulation de ces vésicules dans la région péricanaliculaire est une caractéristique morphologique dans de nombreuses formes de cholestase. L’accumulation des acides biliaires en particulier, l’acide chénodésoxycholique, inhibe la fonction des protéines motrices telles que la kinésine et la dynéine qui permettent à ces vésicules de se mouvoir le long des microtubules et ainsi atteindre le pôle luminal [35] . Un nombre réduit de transporteurs fonctionnels présents à la membrane canaliculaire contribue ainsi à aggraver la cholestase. La signalisation calcium-dépendante dans et entre les hépatocytes est perturbée en cas de cholestase [36]. Les protéines des gap-jonctions (conexine 32 et 36) disparaissent en 24 heures après ligature de la voie biliaire principale chez le rat et est associée à une diminution de la migration des vagues calciques entre les hépatocytes [37]. On pense que ce phénomène est responsable de la diminution du micropéristaltisme canaliculaire qui, normalement, constitue une force motrice facilitant la propulsion de la bile du canalicule vers les ductules biliaires situés dans les espaces porte.
Applications thérapeutiques et perspectives
Le principal traitement des maladies cholestatiques est l’acide ursodésoxycholique le 7bêta épimère de l’acide chénodésoxycholique, un acide biliaire présent en très petite quantité chez l’homme et aux propriétés déconcertantes [39-42]. La base rationnelle de l’utilisation de cet acide biliaire reposait sur l’hypothèse que la cholestase et l’accumulation des acides biliaires hydrophobes dans le foie, du fait de l’inflammation et de la destruction des cholangiocytes étaient en grande partie responsables de l’évolution des maladies cholestatiques vers la cirrhose, et l’insuffisance hépatocellulaire terminale requérant la transplantation hépatique. Cette hypothèse a été testée dans les essais thérapeutiques contrôlés et ils ont montré effectivement que cet acide biliaire particulier stoppait ou ralentissait l’évolution de nombreuses maladies cholestatiques en particulier, la cirrhose biliaire primitive. Ce traitement est de ce fait considéré comme le traitement de référence de la cirrhose biliaire primitive et certaines applications sont étendues à des maladies cholestatiques telles que les cholangites sclérosantes primitives, certaines cholestases familiales progressives, la mucoviscidose, la cholestase gravidique, la cholestase de la nutrition parentérale et certaines cholestases médicamenteuses.
Le mécanisme d’action de l’acide ursodésoxycholique est complexe [43-44]. Cet acide biliaire entraîne une déplétion des acides biliaires endogènes hydrophobes dans le foie et les cholangiocytes, les protégeant ainsi de l’action toxique de ces stéroïdes. En outre, l’acide ursodésoxycholique diminue la réabsorption iléale des acides biliaires. Une des propriétés particulières de l’acide ursodésoxycholique est représentée par ses propriétés anti-apoptotiques [45, 46]. Que celle-ci soit déclenchée par des agents tels que certaines cytokines, les acides biliaires hydrophobes ou certains xénobiotiques. L’acide ursodésoxycholique augmente l’activité canaliculaire des protéines chargées du transport des acides biliaires, de l’hépatocyte vers la bile, en modulant l’activité MAPkinase et protéine kinase C [47, 48]. En outre, l’acide ursodésoxycholique est un agoniste du récepteur nucléaire PXR (Pregnane X Receptor) et de son homologue humain SXR (Steroid Xenobiotic Receptor) [11-14, 49]. D’autres mécanismes pourraient expliquer les effets bénéfiques de l’acide ursodésoxycholique, diminution de l’expression des molécules du complexe majeur d’histocompatibilité sur les hépatocytes et cholangiocytes, augmentation de l’expression et de l’activité de l’échangeur AE2 dans le foie, diminution de l’activité des gènes de l’inflammation (NO synthase, Cox2…) La connaissance de plus en plus précise des transporteurs hépatobiliaires et des récepteurs nucléaires ouvre la voie à la chimiogénomique qui devrait permettre le développement d’agonistes ou d’antagonistes spécifiques des altérations observées au cours des maladies cholestatiques. Ainsi s’ouvre de nouvelles voies thérapeutiques qui devraient diminuer la morbidité, la mortalité de ces maladies invalidantes ainsi que le nombre de transplantations hépatiques réalisées dans ces indications.
BIBLIOGRAPHIE [1] JANSEN P.L. — Foreword : from classic bile physiology to cloned transporters.
Semin Liver Dis 2000, 20 , 245-250.
[2] MEIER P.J., STIEGER B. — Bile salt transporters.
Annu Rev Physiol 2002, 64 , 635-661.
[3] TRAUNER M., MEIER P.J., BOYER J.L. — Molecular pathogenesis of cholestasis.
N Engl J Med 1998 ; 339 , 1217-1227.
[4] CHAUWLA A., REPA J.J., EVANS R.M., MANGELSDORF D.J. — Nuclear receptors and lipid physiology : opening the X-files. Science 2001, 294 , 1866-1870.
[5] OLEFSKY J.M. — A unified nomenclature system for the nuclear receptor superfamily.
Cell 1999, 97 , 161-163.
[6] OFEFSKY J.M. — Nuclear receptor minireview series.
J Biol Chem 2001, 276 , 36863-36864.
[7] KARFEN S.J. — Nuclear receptor regulation of hepatic function.
J Hepatol 2002, 36 , 832-850.
[8] MAKISHIMA M., OKAMOTO A.Y., REPA J.J., TU H., LEARNED R.M., LUL A. — Identification of a nuclear receptor for bile acids. Science 1999, 284 , 1362-1365.
[9] KAST H.R., NGUYEN C.M., SINAL C.J., JONES S.A., LAFFITTE B.A., REUE K. — Farnesoid X-activated receptor induces apolipoprotein C-II transcription : a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol 2001, 15 , 1720-1728.
[10] SINAL C.J., TOHKIN M., MIYATA M., WARD J.M., LAMBERT G., GONZALEZ F.J. — Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102 , 731-744.
[11] XIE W., RADOMINSKA-PANDYA A., SHI Y., SIMON C.M., NELSON M.C., ONG E.S. — An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A 2001, 98 , 3375-3380.
[12] STAUDINGER J., LIU Y., MADAN A., HABEEBU S., KLAASSEN C.D. — Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos 2001, 29 , 1467-1472.
[13] STAUDINGER J.L., GOODWIN B., JONES S.A., HAWKINS-BROWN D., MACKENZIE K.I., LATOUR A.
— The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A 2001, 98 , 3369-3374.
[14] KLIEWER S.A., WILLSON T.M. — Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J Lipid Res 2002, 43 , 359-364.
[15] SYNOLD T.W., DUSSAULT I., FORMAN B.M. — The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med 2001, 7 , 584-590.
[16] SCHUETZ E.G., STROM S., YASUDA K., LECUREUR V., ASSEM M., BRIMER C. — Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J Biol Chem 2001, 276 , 39411-39418.
[17] XIE W., YEUH M.F., RADOMINSKA-PANDYA A., SAINI S.P., NEGISHI Y., BOTTROFF B.S. — Control of steroid, heme, and carcinogen metabolism by nuclear pregnane X receptor and constitutive androstane receptor. Proc Natl Acad Sci U S A 2003 ; 100 , 4150-4155.
[18] FU X., MENKE J.G., CHEN Y., ZHOU G., MACNAUL K.L., WRIGHT S.D. — 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J Biol Chem 2001, 276 , 38378-38387.
[19] PEET D.J., TURLEY S.D., MA W., JANOWSKI B.A., LOBACCARO J.M., HAMMER R.E. — Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93 , 693-704.
[20] SONG C., HIIPAKKA R.A., LIAO S. — Selective activation of liver X receptor alpha by 6alphahydroxy bile acids and analogs. Steroids 2000, 65 , 423-427.
[21] REPA J.J., MANGELSDORF D.J. — The liver X receptor gene team : potential new players in atherosclerosis. Nat Med 2002, 8 , 1243-1248.
[22] WITTENBURG H., CAREY M.C. — Biliary cholesterol secretion by the twinned sterol halftransporters ABCG5 and ABCG8. J Clin Invest 2002, 110 , 605-609.
[23] GRAF G.A., LI W.P., GERARD R.D., GELISSEN I., WHITE A., COHEN J.C. — Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface. J Clin Invest 2002, 110 , 659-669.
[24] YU L., LI-HAWKINS J., HAMMER R.E., BERGE K.E., HORTON J.D., COHEN J.C. — Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest 2002, 110 , 671-680.
[25] JACQUEMIN E., HADCHOUEL M. — Genetic basis of progressive familial intrahepatic cholestasis.
J Hepatol 1999, 31 , 377-381.
[26] JANSEN P.L., MULLER M. — The molecular genetics of familial intrahepatic cholestasis.
Gut 2000, 47 , 1-5.
[27] ROSMORDUC O., HERMELIN B., POUPON R. — MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology 2001, 120 , 1459-1467.
[28] GIRODON E., STERNBERG D., CHAZOUILLERES O., CAZENEUVE C., HUOT D., CALMUS Y. — Cystic fibrosis transmembrane conductance regulator (CFTR) gene defects in patients with sclerosing cholangitis. J Hepatol 2002, 37 , 192-197.
[29] BOHAN A., BOYER J.L. — Mechanisms of hepatic transport of drugs : implications for cholestatic drug reactions. Semin Liver Dis 2002, 22 , 123-136.
[30] LEE J., BOYER J.L. — Molecular alterations in hepatocyte transport mechanisms in acquired cholestatic liver disorders. Semin Liver Dis 2000, 20 , 373-384.
[31] LEE J., AZZAROLI F., WANG L., SOROKA C.J., GIGLIOZZI A., SETCHELL K.D. — Adaptive regulation of bile salt transporters in kidney and liver in obstructive cholestasis in the rat.
Gastroenterology 2001, 121 , 1473-1484.
[32] ZOLLNER G., FICKERT P., ZENZ R., FUCHSBICHLER A., STUMPTNER C., KENNER L. — Hepatobiliary transporter expression in percutaneous liver biopsies of patients with cholestatic liver diseases. Hepatology 2001, 33 , 633-646.
[33] PHILLIPS M.J., POUCELL S., ODA M. — Mechanisms of cholestasis.
Lab Invest 1986, 54 , 593-608.
[34] FALLON M.B., BRECHER A.R. BALDA M.S., MATTER K. ANDERSON J.M. — Altered hepatic localization and expression of occludin after common bile duct ligation. Am J Physiol 1995, 269 , C1057-C1062.
[35] MARKS D.L., LARUSSO N.F., MC NIVEN M.A. — Isolation of the microtubule-vesicle motor kinesin from rat liver : Selective inhibition by cholestatic bile acids. Gastroenterology 1995, 108 , 824-833.
[36] BEUERS U., NATHANSON M.H., ISALES C.M., BOYER J.L. — Tauroursodeoxycholic acid stimulates hepatocellular exocytosis and mobilizes extracellular Ca++ mechanisms defective in cholestasis. J Clin Invest 1993, 92 , 2984-2993.
[37] FALLON M.B., NATHANSON M.H., MENNONE A., SAEZ J.C., BURGSTAHLER A.D., ANDERSON J.M. — Altered expression and function of hepatocyte gap junctions after common bile duct ligation in the rat. Am J Physiol 1995, 268 , C1186-C1194.
[38] POUPON R., CHRÉTIEN Y., POUPON R.E., BALLET F., CALMUS Y., DARNIS F. — ‘‘Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis ? ’’ Lancet 1987, 8537 , 834-836.
[39] POUPON R.E., BALKAU B., ESCHWEGE E., POUPON R. — A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC study Group. N Engl J Med , 1991, 324 , 1548-1854.
[40] POUPON R.E., POUPON R., BALKAU B. — Ursodiol for the long-term treatment of primary biliary cirrhosis. The UDCA-PBC Study Group. N Engl J Med 1994, 330 , 1342-1347.
[41] POUPON R.E., LINDOR K.D., CAUCH-DUDEK K., DICKSON E.R., POUPON R., HEATHCOTE E.J. — Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology 1997, 113 , 884-890.
[42] POUPON R., POUPON R.E. — Ursodeoxycholic acid therapy of chronic cholestatic conditions in adults and children. Pharmacol Ther 1995 ; 66 , 1-15.
[43] PAUMARTNER G., BEUERS U. — Ursodeoxycholic acid in cholestatic liver disease : mechanisms of action and therapeutic use revisited. Hepatology 2002, 36 , 525-531.
[44] RODRIGUES C.M., FAN G., WONG P.Y., KREN B.T, STEER C.J. — Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med 1998 ; 4 , 165-178.
[45] RODRIGUES C.M., FAN G., MA X., KREN B.T., STEER C.J. — A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest 1998 ; 101 , 2790-2799.
[46] KURZ A.K., GRAF D., SCHMITT M., VOM DAHL S., HAUSSINGER D. — Tauroursodesoxycholateinduced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology 2001, 121 , 407-419.
[47] BEUERS U., BILZER M., CHITTATTU A., KULLAK-UBLICK G.A., KEPPLER D., PAUMGARTNER G.
— Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C-dependent mechanisms in cholestatic rat liver. Hepatology 2001, 33 , 1206-1216.
[48] SONODA J., XIE W., ROSENFELD J.M., BARWICK J.L., GUZELIAN P.S., EVANS R.M. — Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR). Proc Natl Acad Sci U S A 2002, 99 , 13801-13806.
[49] WEI P., ZHANG J., EGAN-HAGLEY M., LIANG S., MOORE D.D. — The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 2000, 407, 920-3.
[50] ZHANG J., HUANG W., CHUA S.S., MOORE D.D. — Modulation of acetaminophen toxicity by the xenobiotic receptor CAR. Science 2002, 298 : 422-4.
[51] HUANG W., ZANG J., CHUA S.S., QATANI M., HAN Y., GREANATA R., MOORE D.D. — Induction of bilirubin clearance by the constitutive androstane receptor (CAR). PNAS 2003, 100, 4156-61.
DISCUSSION
M. Michel BOUREL
A propos de l’acide ursodésoxycholique : Quelle responsabilité dans l’émergence du cancer colorectal ? Quelle nécessité d’y ajouter d’autres molécules et dans quelles circonstances ? A propos des transporteurs membranaires, quel mode de transmission familiale des cholestases progressives familiales ?
L’administration orale prolongée d’acide ursodésoxycholique diminue l’absorption iléale des acides biliaires endogènes, l’acide cholique et chénodésoxycholique. En outre,
l’acide ursodésoxycholique est en partie métabolisé par la flore intestinale dans le colon en acides chénodésoxycholique et lithocholique. Il existe des arguments épidémiologiques et expérimentaux suggérant le rôle des acides biliaires dans la carcinogénèse colorectale. En d’autres termes, lorsque l’acide ursodésoxycholique a été introduit en thérapeutique humaine, on pouvait craindre la possibilité de survenue plus fréquente d’adénomes ou de cancers colorectaux. Cependant, très rapidement il a été montré que l’acide ursodésoxycholique diminuait de façon marquée la carcinogénèse colorectale chimio-induite. Trois études publiées entre 2001 et 2003 montrent que le traitement prolongé par l’acide ursodésoxycholique diminue significativement l’incidence des dysplasies sévères et des cancers colorectaux au cours de la cholangite sclérosante et diminue la récidive des adénomes colorectaux au cours de la cirrhose biliaire primitive. Les études préliminaires montrent également que l’effet anti-carcinogène de l’acide ursodésoxycholique pourrait se manifester également vis-à-vis du cholangiocarcinome, — complication fréquente des cholangites sclérosantes primitives. S’agissant du traitement de la cirrhose biliaire primitive, l’acide ursodésoxycholique entraine une rémission de cette maladie dans 30 à 40 % des cas et une progression de la survie sans transplantation. Cependant, certaines formes très actives de la maladie peuvent évoluer vers l’insuffisance hépatique et le recours à la transplantation est nécessaire. Dans ces formes, il est souvent nécessaire d’associer la Prednisone voire l’Azathiopine et le Mycophénolate Mofétil. Les cholestases familiales constituent un groupe de maladies rares autosomales récessives dont l’incidence est estimée à 1/100 000. Les cholestases familiales de type 1 et 2 représentent 2/3 des cas et les cholestases familiales de type 3 1/3 des cas.
M. Raymond ARDAILLOU
Quel est le rôle des enzymes intervenant dans la synthèse des acides biliaires ? Leur dysfonctionnement est-il impliqué en pathologie hépatique ? Quels sont les facteurs contrô- lant la progression de la sclérose hépatique au cours des maladies biliaires ?
La synthèse des acides biliaires fait intervenir une quinzaine d’enzymes, principalement des cytochromes, localisés dans les hépatocytes. Certaines formes très rares de cholestase familiale progressive relèvent de l’absence d’une de ces enzymes. Le diagnostic ne peut être fait que par une analyse chromatographique et en spectrométrie de masse des acides biliaires dans le sang, ou mieux les urines de ces enfants. Jusqu’à présent, il n’a pas été mis en évidence chez l’adulte jeune de pathologie biliaire relevant d’anomalies enzymatiques de la synthèse des acides biliaires. Cependant, il est probable qu’une analyse systématique des cholestases inexpliquées observées chez l’adulte jeune pourrait mettre en évidence de telles anomalies. Les facteurs contrôlant la progression de la fibrose hépatique au cours des maladies biliaires font l’objet d’études expérimentales dans notre unité INSERM.
Deux types cellulaires sont activés au cours des maladies biliaires, en premier lieu les fibroblastes juxtacanalaires des espaces portes et d’autre part les cellules étoilées présentes dans le sinusoïde hépatique. L’un des médiateurs mis en jeu ayant une importance semble-t-il capitale est le PDGF et son récepteur thyrosine kinase. Un autre mécanisme mis en jeu est l’ischémie et l’hypoxie. En effet, au cours des maladies biliaires, l’inflammation entraine des lésions artériolaires et veinulaires responsables d’un défaut de perfusion hépatique, responsable lui-même d’un remodelage parenchymateux associé à une activation des cellules impliquées dans la synthèse du collagène. Comme en pathologie rénale ou d’autres circonstances, la connaissance des mécanismes biologiques de la fibrose et de son développement a des implications importantes pour les traitements futurs.
M. Alain LARCAN
Pouvez-vous préciser les mécanismes en cause susceptibles d’expliquer les cholestases gravidiques récidivantes et de la cholestase récurrente bénigne ? Quels sont les mécanismes en cause dans les cholestases après ischémie ou pertrubations microcirculatoires .
Les cholestases gravidiques se caractérisent par la survenue de cholestase (prurit, élévation marquée des acides biliaires sériques) survenant dans le dernier trimestre de la grossesse, et pouvant aboutir à la mort in utero du fœtus ou à une prématurité. Les cholestases gravidiques sont en partie dues à des anomalies des gènes responsables de la synthèse des protéines de transport des acides biliaires dans le foie. C’est ainsi que les mutations du gène MDR3 et de BSEP ont pu être mises en évidence dans certaines cholestases gravidiques. D’autres cholestases gravidiques pourraient être dues à des anomalies du transport des métabolites de la progestérone et des oestrogènes avec comme conséquence un arrêt de la sécrétion biliaire. La cholestase récurrente bénigne (« maladie de Summerskill ») est une entité clinique au cours de laquelle des mutations faux sens du gène PFIC1 (le même gène en cause dans la maladie de Byler ou cholestase failiale de type 1 du nouveau-né). Au cours des états de choc et des hépatites ischémiques se développe fréquemment une cholestase marquée. Celle-ci pourrait être due à l’effet conjoint de l’hypoxie, de l’ischémie et des effets des endotoxines. En effet, la combinaison d’ischémie et d’endotoxinémie entraine expérimentalement, non seulement une nécrose hépatique mais une cholestase marquée. Dans ces circonstances, il est en effet mis en évidence une diminution de la quantité et de l’activité des transporteurs canaliculaires responsables de la sécrétion canaliculaire de la bilirubine, du glutathion et des acides biliaires.
* Chef du Service d’Hépatologie, Hôpital Saint-Antoine, AP Hôpitaux de Paris 184, rue du Faubourg Saint-Antoine — 75571 Paris Cedex 12 Professeur des Universités, UFR Saint-Antoine, Univ. Paris 6 INSERM U.402 Tirés-à-part : Professeur POUPON à l’adresse ci-dessus. Article reçu le 22 avril 2003, accepté le 2 juin 2003 .
Bull. Acad. Natle Méd., 2003, 187, no 7, 1261-1276, séance du 21 octobre 2003