
Résumé
La maladie d’Alzheimer est caractérisée par la présence, de deux types de lésions dans le cerveau : les dégénérescences neurofibrillaires et les plaques séniles. La coexistence de ces deux lésions est indispensable pour confirmer le diagnostic clinique de la maladie d’Alzheimer. Leur caractérisation biochimique a permis d’identifier les protéines majeures qui les composent. Les dégénérescences neurofibrillaires contiennent la protéine tau hyperphosphorylée, et les plaques séniles un noyau de peptide amyloïde provenant du métabolisme de son précurseur. L’analyse biochimique de ces protéines dans des modèles cellulaires spécifiques a permis de montrer que la phosphorylation du précurseur du peptide amyloïde et de la protéine tau constitue un lien biochimique entre les deux lésions caractéristiques de la maladie d’Alzheimer. Dans les neurones, cette phosphorylation est réalisée par des protéines kinases qui peuvent être activées par le calcium. Dès lors, il est important de contrôler l’homéostasie calcique neuronale et d’éviter toute augmentation de la concentration cytosolique en calcium.
Summary
Alzheimer’s disease is characterized by the presence of neurofibrillary tangles and senile plaque in the brain. Both disorders must be present in order to confirm a clinical diagnosis of Alzheimer’s disease. Neurofibrillary tangles contain hyperphosphorylated microtubuleassociated protein tau, while senile plaque contains a core of amyloid peptide derived from its precursor. Phosphorylation of both amyloid precursor protein and tau represents a biochemical link between the two characteristic lesions of Alzheimer’s disease.
LES PROTÉINES DES LÉSIONS CARACTÉRISTIQUES DE LA MALADIE D’ALZHEIMER
La protéine tau hyperphosphorylée des dégénérescences neurofibrillaires
L’examen neuropathologique du cerveau de patients atteints de maladie d’Alzheimer révèle deux types de lésions caractéristiques : les dégénérescences neurofibrillaires et les plaques séniles. Les premières sont constituées de paires hélicoïdales de filaments contenant la protéine tau [1]. Celle-ci est une protéine neuronale associée aux microtubules. Tau joue un rôle essentiel dans le maintien du réseau microtubulaire utilisé par le neurone pour le transport vésiculaire du flux axoplasmique. Le gène de la protéine tau est localisé sur le chromosome 17 humain. Par épissage alternatif, ce gène donne naissance à différents ARN messagers traduits en six protéines différentes. En conditions physiologiques, les protéines tau sont des phospho-protéines solubles. En revanche, dans le cerveau de patients atteints de la maladie d’Alzheimer, les protéines tau s’organisent en paires hélicoïdales de filaments insolubles. L’analyse biochimique montre que les protéines tau du cerveau pathologique sont hyperphosphorylées sur de nombreux résidus sérines et thréonines [2].
La glycogène synthase kinase 3 β (GSK3β) est capable de phosphoryler la majorité des résidus sérines et thréonines des protéines tau hyperphosphorylées[3]. D’autres kinases phosphorylent aussi ces sites, notamment la Cycline-dépendante kinase 5 (Cdk5). Les protéines tau hyperphosphorylées non seulement dissocient les microtubules et ne sont donc plus capables de les stabiliser, mais de plus s’organisent en paires hélicoïdales de filaments. Ceci a pour conséquence une perturbation très importante du flux axoplasmique, à un point tel qu’un neurone contenant des dégénérescences neurofibrillaires est destiné à mourir. Il existe d’ailleurs une excellente corrélation entre le nombre de dégénérescences neurofibrillaires dans certaines régions du cerveau et la sévérité du syndrome démentiel.
Le précurseur du peptide amyloïde des plaques séniles
Les plaques séniles sont des lésions sphériques extracellulaires contenant un noyau de substance amyloïde dont le constituant majeur est le peptide Aβ[4]. Ce peptide amyloïde, de 39-43 acides aminés, provient d’un précurseur plus grand : le précurseur du peptide amyloïde ou APP (Amyloid Precursor Protein) [5]. Le gène de l’APP est localisé sur le chromosome 21 humain. L’épissage alternatif de ce gène fournit différents ARN messagers qui sont traduits en dix isoformes de l’APP contenant de 365 à 770 acides aminés. Les neurones synthétisent essentiellement l’APP contenant 695 acides aminés (APP695). Cette protéine est une protéine membranaire qui ne comporte qu’un seul domaine transmembranaire (Figure 1). La séquence du peptide amyloïde Aβ est partiellement transmembranaire et partiellement extracellulaire (Figure 1). L’analyse biochimique de l’APP a permis d’identifier différents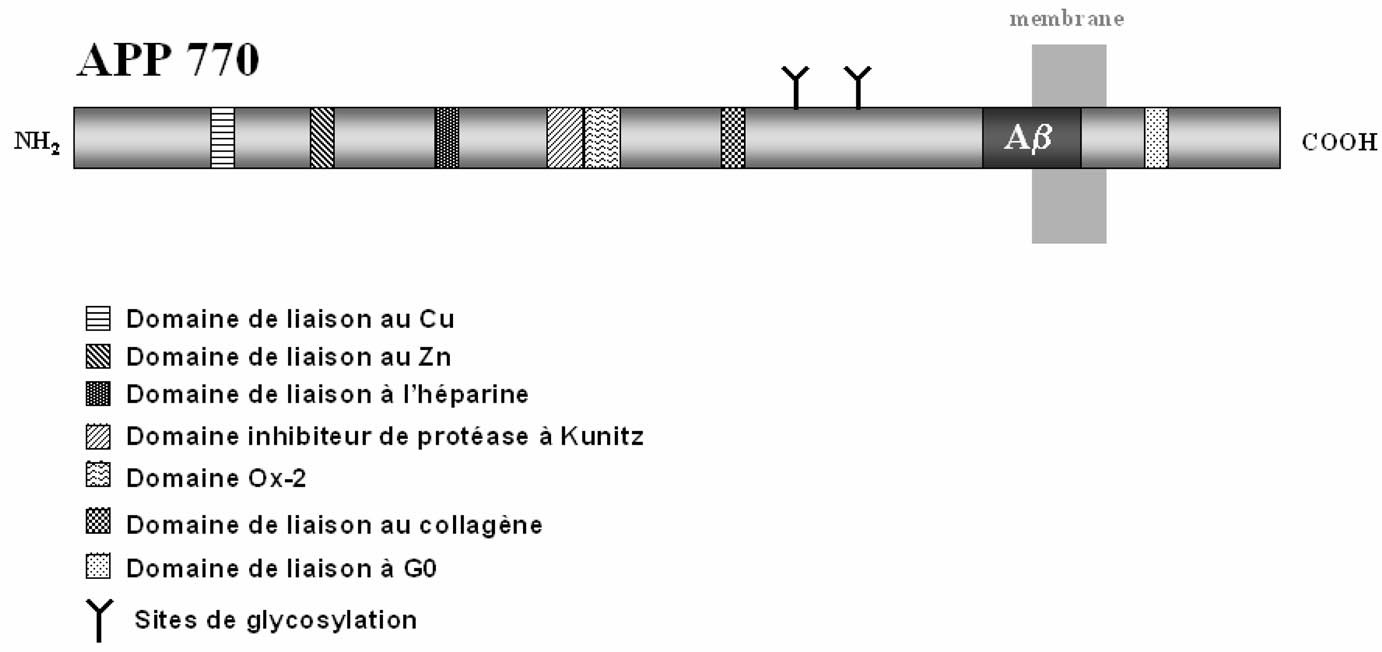
FIGURE 1. — Représentation schématique de la structure et des domaines fonctionnels de l’APP770.
domaines de la protéine. Certaines isoformes de l’APP sont des inhibiteurs de protéases à sérine et le domaine intracytoplasmique de l’APP est capable d’interagir avec la protéine Go (Figure 1). Ce même domaine intracytoplasmique est capable aussi d’interagir avec des protéines adaptatrices, telle la protéine Fe65. L’APP est capable d’interagir avec des ions tels que le Cu++ ou le Zn++, mais aussi avec l’héparine, les héparanes sulfates protéoglycans, ou avec des protéases (Figure 1).
L’identification de ces différents domaines fait de l’APP une protéine particulièrement polyvalente, mais sa fonction essentielle reste inconnue [6].
LE MÉTABOLISME CELLULAIRE DU PRÉCURSEUR DU PEPTIDE AMYLOIDE
La voie catabolique non amyloïdogène du précurseur du peptide amyloïde
L’expression de l’APP par transfection cellulaire des ADN complémentaires correspondants et l’utilisation d’anticorps reconnaissant spécifiquement la protéine ont permis d’étudier le métabolisme de l’APP. Les cellules transfectées exprimant l’APP libèrent de la protéine soluble dans le milieu de culture. Celle-ci est reconnue par des anticorps dirigés contre la partie N-terminale de l’APP, mais non par des anticorps dirigés contre la partie C-terminale de la protéine. L’APP soluble extracellulaire est obtenue par clivage de la protéine transmembranaire au sein même de la séquence du peptide Aβ [7]. Ce clivage est réalisé par une activité α-sécrétase.
L’α-sécrétase ne clive pas spécifiquement une séquence en acides aminés ; en revanche, elle est très sensible à la distance qui sépare le site de clivage du domaine transmembranaire de l’APP. Cette distance ne doit pas dépasser douze à treize acides aminés [8]. Des inhibiteurs de protéases, ajoutés au milieu extracellulaire de cellules exprimant l’APP, n’empêchent pas la production d’APP soluble. Ceci suggère que le clivage effectué par l’α-sécrétase a lieu lors de l’acheminent intracellulaire de l’APP vers la membrane plasmique. Des protéases de la famille des désintégrines et métallo-protéases, telles ADMA10 et ADAM17, sont sans doute impliquées dans le clivage α-sécrétase de l’APP [9,10]. Le clivage de l’APP au sein même de la séquence du peptide Aβ empêche la formation de peptide amyloïde intact, celui qui se dépose dans le noyau amyloïde des plaques séniles. Cette voie catabolique de l’APP est donc appelée voie catabolique non amyloïdogène (Figure 2).
suggère que le clivage effectué par l’α-sécrétase a lieu lors de l’acheminent intracellulaire de l’APP vers la membrane plasmique. Des protéases de la famille des désintégrines et métallo-protéases, telles ADMA10 et ADAM17, sont sans doute impliquées dans le clivage α-sécrétase de l’APP [9,10]. Le clivage de l’APP au sein même de la séquence du peptide Aβ empêche la formation de peptide amyloïde intact, celui qui se dépose dans le noyau amyloïde des plaques séniles. Cette voie catabolique de l’APP est donc appelée voie catabolique non amyloïdogène (Figure 2).
FIG. 2. — Les deux voies cataboliques de l’APP. La voie catabolique non amyloïdogène : clivage par l’α-sécrétase. La voie catabolique amyloïdogène : clivage par les β- et g-sécrétases.
La voie catabolique amyloïdogène du précurseur du peptide amyloïde
Parallèlement à la voie catabolique non amyloïdogène, les cellules transfectées qui expriment l’APP sont capables de produire du peptide amyloïde soluble extracellulaire[11]. Cela implique d’une part l’inhibition de l’activité α-sécrétase et, d’autre part, l’activation de deux autres protéases qui clivent l’APP et libérent le peptide Aβ.
Du côté N-terminal du peptide amyloïde, le clivage est réalisé par une activité β-sécrétase. Cette activité est portée par la protéine BACE, une aspartyl protéase membranaire[12]. Ce clivage génère un fragment C-terminal de l’APP qui contient l’ensemble du peptide Aβ. Les fragments C-terminaux amyloïdogènes sont ensuite clivés par une activité g-sécrétase. Cette activité est portée par un complexe multiprotéique contenant au moins 4 protéines différentes, dont une préséniline qui joue un rôle essentiel dans ce clivage[13]. Ce double clivage de l’APP libère le peptide amyloïde intact dans le milieu extracellulaire (Figure 2).
L’APP utilise donc deux voies cataboliques distinctes (Figure 2) : une voie non amyloïdogène au cours de laquelle la protéine transmembranaire est clivée au sein du peptide amyloïde, la région N-terminale de la protéine étant ensuite libérée dans le milieu extracellulaire, et une voie catabolique amyloïdogène aboutissant à la sécrétion de peptide amyloïde soluble. Cette dernière voie catabolique, largement minoritaire, ne représente qu’un très faible pourcentage du métabolisme de l’APP.
Le peptide amyloïde n’est pourtant pas un produit de dégradation anormal de l’APP.
NEUROTOXICITÉ DU PEPTIDE AMYLOIDE
Le peptide amyloïde soluble extracellulaire n’est pas toxique
Les cellules transfectées qui expriment l’APP humain ont fourni de nombreux renseignements sur le métabolisme de l’APP. Ces cellules produisent du peptide amyloïde à partir de son précurseur et ne montrent pourtant souvent aucun signe de toxicité. Il faut pourtant noter que les cellules transfectées de type fibroblastique largement utilisées sont fort différentes des neurones qui synthétisent l’APP cérébral et dégénèrent dans la maladie d’Alzheimer. La culture primaire de neurones embryonnaires permet d’étudier le métabolisme neuronal des protéines. L’utilisation de neurones d’origine humaine nécessite de disposer de cerveau embryonnaire.
Pour des raisons éthiques, nous avons utilisé, comme d’autres, des neurones d’embryons de rat ou de souris. Ces neurones de rongeurs non transfectés produisent de l’APP endogène, mais très peu de peptide amyloïde puisqu’ils utilisent essentiellement la voie catabolique non amyloïdogène de l’APP (alpha-sécrétase).
C’est sans doute la raison pour laquelle les rongeurs ne développent pas de plaques séniles. Ceci est probablement expliqué par de légères différences de séquence au niveau du peptide amyloïde entre les APP humains et de rongeurs. Pour exprimer l’APP humain dans des cultures primaires de neurones de rats ou de souris, nous avons utilisé des vecteurs d’origine virale [14]. Ceux-ci permettent l’expression de protéines humaines dans des cultures primaires de neurones de rats. Il devient donc possible d’étudier le métabolisme de l’APP humain dans les neurones, cellules qui sont directement impliquées dans le développement de la maladie d’Alzheimer.
Dans ces conditions, l’expression d’APP humain dans les neurones de rat en culture induit une importante neurotoxicité, contrairement à ce qui était observé dans des cellules transfectées. Néanmoins, le peptide amyloïde soluble produit dans le milieu de culture de neurones ou de cellules transfectées n’induit aucune toxicité. Le peptide amyloïde extracellulaire ne devient toxique pour les neurones lorsqu’il forme des oligomères ou des structures fibrillaires. La neurotoxicité induite par l’expression neuronale de l’APP n’est donc pas liée à la production de peptide amyloïde soluble extracellulaire [15].
Le peptide amyloïde intraneuronal est neurotoxique
Contrairement aux cellules fibroblastiques transfectées, les neurones produisent des quantités détectables de peptide amyloïde intracellulaire. Dès l’apparition de peptide Aβ intracellulaire, leur survie diminue de façon très significative [15]. De plus, un inhibiteur fonctionnel de la g-sécrétase permet de réduire de manière importante la production de peptide Aβ intraneuronal et protège les neurones de la toxicité du peptide Aβ. Cette toxicité résulte de la formation, à l’intérieur des neurones, de trimères et tétramères d’Aβ. Ces petits oligomères sont à l’origine d’une neurotoxicité importante [16]. Dans différents modèles expérimentaux et chez les patients atteints de maladie d’Alzheimer, l’accumulation de peptide amyloïde intraneuronal parait être un événement précoce, à l’origine d’une perte neuronale significative.
HOMEOSTASIE CALCIQUE ET MÉTABOLISME DU PRÉCURSEUR DU PEPTIDE AMYLOIDE
Une augmentation de la concentration cytosolique en calcium provoque l’accumulation de peptide amyloïde intraneuronal
Dans tout type cellulaire, et particulièrement les neurones, il est essentiel de contrôler l’homéostasie calcique de manière stricte et rigoureuse. Dans des neurones en culture, une modification de la concentration cytosolique en calcium peut être provoquée par l’ouverture des canaux calciques dépendants du voltage, après dépolarisation par du potassium. Dans ces conditions expérimentales, une accumulation importante de peptide Aβ intraneuronal est mesurée [17]. Cette accumulation d’Aβ intraneuronal est précédée par la phosphorylation transitoire de l’APP695 sur la thréonine 668 [18]. Cette phosphorylation est indispensable à l’accumulation de peptide amyloïde intraneuronal.
La phosphorylation : un lien biochimique entre les plaques séniles et les dégénérescences neurofibrillaires
À la suite de l’élévation de la concentration cytosolique en calcium, l’activation des protéines kinases GSK3β et Cdk5 augmente non seulement la phosphorylation de l’APP695 sur la thréonine 668 mais induit également la phosphorylation transitoire de la protéine tau sur les résidus serines 396 et 404. Dans les paires hélicoïdales de filaments, la protéine tau est phosphorylée sur ces résidus. Dès lors, la phosphorylation des protéines tau et APP dans des neurones en culture, résultant d’une modification de la concentration cytosolique en calcium, est capable d’induire des modifications biochimiques de deux protéines impliquées dans le développement de la maladie d’Alzheimer. En effet, la protéine tau hyperphosphorylée est le constituant majeur des dégénérescences neurofibrillaires, alors que la phosphorylation de l’APP conduit à la production de peptide Aβ des plaques séniles.

FIG. 3. — La phosphorylation des protéines neuronales tau et APP, résultant d’une augmentation de la concentration cytosolique en calcium, constitue un lien biochimique entre les lésions caractéristiques de la maladie d’Alzheimer.
Le vieillissement est le facteur de risque essentiel associé au développement de la maladie d’Alzheimer. Au cours du vieillissement, une défaillance des mécanismes cellulaires régulant l’homéostasie calcique est souvent constatée. De plus, l’inflammation, l’excito-toxicité et le stress oxydatant associés au vieillissement cérébral contribuent eux aussi à augmenter la concentration cytosolique en calcium neuronal. Dans ces conditions, des protéines kinases telles que la GSK3β et la Cdk5 sont activées et phosphorylent différentes protéines dont les protéines tau et APP, conduisant à l’accumulation intraneuronale de protéine tau hyperphospho-
rylée et de peptide Aβ (Figure 3). Une phosphorylation anormale, liée à une perturbation de l’homéostasie calcique permet donc d’établir un lien biochimique entre les dégénérescences neurofibrillaires et les plaques séniles de la maladie d’Alzheimer. Dès lors, la poursuite du développement de molécules capables de contrôler pharmacologiquement la concentration cytosolique en calcium neuronal pourrait constituer une approche thérapeutique intéressante pour le traitement de la maladie d’Alzheimer.
BIBLIOGRAPHIE [1] BRION J.P., COUCK A.M., PASSAREIRO E., FLAMENT-DURAND J. — Neurofibrillary tangles of Alzheimer’s disease : an immunohistochemical study. J. Submicrosc. Cytol., 1985, 17 , 89-96.
[2] REYNOLDS C.H., BETTS J.C., BLACKSTOCK W.P., NEBREDA A.R. & ANDERTON B.H. — Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry : differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta.
J. Neurochem., 2000, 74 , 1587-1595.
[3] HANGER D.P., HUGHES K., WOODGETT J.R., BRION J.P., ANDERTON B.H. — Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau : generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett., 1992, 147 , 58-62.
[4] GLENNER G.G., WONG C.W. — Alzheimer’s disease : initial report of the purification and characterization of a novel cerebrovascular amyloid protein Biochem. Biophys. Res. Commun., 1984, 120 , 885-890.
[5] KANG J., LEMAIRE H.G., UNTERBECK A., SALBAUM J.M., MASTERS C.L., GRZESCHIK K.H., MULTHAUP G., BEYREUTHER K., MULLER HILL B. — The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature, 1987, 325 , 733-736.
[6] OCTAVE J.N. — The amyloid peptide precursor in Alzheimer’s disease.
Rev. Neurosci. , 1995, 6 , 287-316.
[7] SCH F.S., KEIM P.S., BEATTIE E.C., BLACHER R.W., CULWELL A.R., OLTERSDORF T., MCCLURE D., WARD P.J. — Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science, 1990, 248 , 1122-1124.
[8] SISODIA S.S. — Beta-amyloid precursor protein cleavage by a membrane-bound protease . Proc.
Natl. Acad. Sci. U.S.A., 1992, 89 , 6075-6079.
[9] LAMMICH S., KOJRO E., POSTINA R., GILBERT S., PFEIFFER R., JASIONOWSKI M., HAASS C., AHRENHOLZ F. — Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U. S. A, 1999, 96 , 3922-3927.
[10] BUXBAUM J.D., LIU K.N., LUO Y., SLACK J.L., STOCKING K.L., PESCHON J.J., JOHNSON R.S., CASTNER B.J., CERRETTI D.P.,BLACK R.A. — Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. , 1998, 273 , 27765-27767.
[11] HAASS C., SCHLOSSMACHER G., HUNG A.Y., VIGO-PELFREY C., MELLON A., OSTASZEWSKI B.L., LIEBERBURG I., KOO E.H., SCHENK D., TEPLOW D.B. — Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature , 1992, 359 , 322-325.
[12] VASSAR R., BENNETT B.D., BABU-KHAN S., KAHN S., MENDIAZ E.A., DENIS P., TEPLOW D.B., ROSS S., AMARANTE P., LOELOFF R., LUO Y., FISHER S., FULLER J., EDENSON S., LILE J., JAROSINSKI M.A., BIERE A.L., CURRAN E., BURGESS T., LOUIS J.C., COLLINS F., TREANOR J., ROGERS G., CITRON M. — Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE [see comments], Science , 1999, 286 , 735-741.
[13] DE STROOPER B. — Aph-1, Pen-2, and Nicastrin with Presenilin Generate an Active gammaSecretase Complex. Neuron , 2003, 38 , 9-12.
[14] MACQ A.F., CZECH C., ESSALMANI R., BRION J.P., MARON A., MERCKEN L., PRADIER L., OCTAVE J.N. — The long term adenoviral expression of the human amyloid precursor protein shows different secretase activities in at cortical neurons and astrocytes. J. Biol. Chem ., 1998, 273 , 28931-28936.
[15] KIENLEN-CAMPARD P., MIOLET S., TASIAUX B., OCTAVE J.N. — Intracellular Amyloid-beta 1-42, but Not Extracellular Soluble Amyloid- beta Peptides Induces Neuronal Apoptosis. J. Biol.
Chem., 2002, 277 , 15666-15670.
[16] PIKE C.J., WALENCEWICZ A.J., GLABE C.G., COTMAN C.W. — Aggregation-related toxicity of synthetic beta-amyloid protein in hippocampal cultures. Eur. J. Pharmacol., 1991, 207 , 367-368.
[17] PIERROT N., GHISDAL P., CAUMONT A.S., OCTAVE J.N. — Intraneuronal amyloid-beta1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J. Neurochem. , 2004, 88 , 1140-1150.
[18] PIERROT N., SANTOS S.F., FEYT C., MOREL M., BRION J.P., OCTAVE J.N. — Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J. Biol. Chem., 2006, 281 , 39907-39914.
DISCUSSION
M. Bernard PESSAC
Quelle(s) est(sont) la(es) relation(s) entre le peptide neurotoxique et le type neuronal affecté ?
Dans le modèle cellulaire utilisé, les neurones en culture sont des neurones corticaux de rat qui présentent une grande hétérogénéité. Un toxicité du peptide amyloïde a été également observée dans des cultures primaires de neurones hippocampe.
M. Pierre DELAVEAU
A-t-on des connaissances précises sur la phosphorylation excessive de la protéine Tau et les liaisons phosphoryle sont-elles riches en énergie ?
La protéine tau est une phosphoprotéine dont de nombreux résidus sérine et thréonine sont phosphorylés en condition physiologique. Dans la maladie d’Alzheimer, c’est le nombre de résidus sérine et thréonine phosphorylés qui augmente, en particulier à proximité du domaine de tau qui interagit avec la tubuline. Les liaisons entre les groupements phosphates et les acides aminés phosphorylables sont de haute énergie, leur formation et leur hydrolyse sont catalysées par des protéine kinases et des protéine phosphatases, respectivement.
M. Jean-Luc de GENNES
Pourquoi dans les trois premières présentations, n’a-t-il pas été fait allusion à la contribution, dans les mécanismes histologiques, des dépôts amyloïdes, des troubles métaboliques lipidiques ? Alors que la participation d’un isomère de l’ApoE, l’ApoE4 chez l’homme double la fréquence, à l’état hétérozygote, de la maladie d’Alzheimer tandis que les porteurs d’ApoE2 et ApoE3 diminuent nettement cette fréquence. En plus des mutations de l’adénosine triphosphate cassettes de liaison telles le SVP, celle de l’ABCK1 et ApoBCG1 sont nettement mises en cause dans les troubles de la décharge extracellulaire du polypeptide A β .
Il existe effectivement un polymorphisme du gène codant l’apolipoprotéine E, à l’origine de l’existence de quatre allèles différents. Les personnes qui sont homozygotes pour l’allèle e4 du gène de l’ApoE ont environ dix fois plus de risque de développer la maladie d’Alzheimer. Étant donné que l’ApoE est de loin l’apolipoprotéine la plus abondante dans le cerveau, ceci indique clairement une relation entre la maladie d’Alzheimer et le métabolisme lipidique. De plus, les statines semblent capables de réduire la production de peptide amyloïde. Les mécanismes cellulaires à l’origine de ces observations doivent encore être élucidés.
M. Michel BOUREL
Quels sont les apports physiopathologiques du fait de l’utilisation des cocultures (neurones, astrocytes, éventuellement cellules somatiques… ?), en dehors de l’allongement du temps d’observation ?
L’avantage des cocultures de neurones et d’astrocytes ou de cellules microgliales est de pouvoir étudier l’influence de cellules gliales sur le métabolisme neuronal de protéines impliquées dans la maladie d’Alzheimer. De plus, on constate dans de nombreuses maladies neurodégénératives la présence d’une réaction de type inflammatoire, dans laquelle les cellules gliales jouent un rôle essentiel.
M. Alain LARCAN
A la recherche des marqueurs biologiques sensibles et spécifiques, avez-vous connaissance de recherches récentes concernant le profil du transcriptome, portant sur les protéines du signal, c’est-à-dire des communications inter-cellulaires ? Je crois avoir lu que le profil de l’Alzheimer est caractéristique et différent de celui du Parkinson.
L’analyse du transcriptome permet d’étudier, en une seule expérience, une différence de transcription de plusieurs milliers de gènes dans deux conditions expérimentales diffé- rentes. En particulier, on peut comparer un tissu contrôle avec un tissu pathologique.
Cette méthode, extrêmement puissante, demande de nombreuses confirmations avant de pouvoir identifier des marqueurs spécifiques et sensibles. Une analyse des protéines (protéomique) entre deux échantillons permet également de dresser un profil de marqueurs à usage diagnostic. Ces méthodes permettront sans doute d’identifier un profil de marqueurs biochimiques utile au diagnostic de la maladie d’Alzheimer.
* Laboratoire de Pharmacologie Expérimentale, Université catholique de Louvain, FARL 5410, Avenue Hippocrate 54 B-1200 Bruxelles. Tirés-à-part : Professeur Jean-Noël OCTAVE, même adresse. Article reçu et accepté le 11 février 2008.
Bull. Acad. Natle Méd., 2008, 192, no 2, 323-332, séance du 12 février 2008