
Résumé
Le cadre des anomalies héréditaires du métabolisme du fer s’est récemment élargi et diversifié. Le tableau de l’hémochromatose génétique correspond aujourd’hui à six entités distinctes ; à l’hémochromatose classique HFE 1 sont en effet venues s’ajouter : l’hémochromatose juvénile HFE 2 dont le gène localisé sur le chromosome 1 n’a pas encore été identifié, HFE 3 par anomalie du deuxième récepteur de la transferrine TfR2, HFE 4 par mutation de la ferroportine (ou SLC 11A3), HFE 5 due à des mutations des chaînes H de la ferritine et HFE 6 dont le gène est celui de l’hepcidine (HAMP). Des surcharges martiales généralisées s’observent également dans l’acéruloplasminémie, l’atransferrinémie et au cours du syndrome « Gracile » par mutation du gène BCS1L. Enfin à cette liste il convient d’ajouter l’hémochromatose néo-natale et la surcharge ferrique des Africains dont les pathogénies sont encore obscures. La physiopathologie d’autres affections génétiques à symptomatologie plus ciblée entraîne des surcharges localisées en fer : ataxie de Friedreich par extension de triplets dans le gène de la frataxine, deux anémies sidéroblastiques à transmission liée au chromosome X par mutations de la δ amino-lévulinate synthétase (ALAS2) et du gène ABC-7, syndrome d’Hallervorden Spatz dont le gène est la panthothénate kinase 2 (PANK2), neuroferritinopathie par mutation de la L-ferritine et enfin syndrome hyperferritinémie-cataracte également due à des mutations de la L-ferritine. Deux autres caractéristiques ajoutent à la diversité des anomalies génétiques du métabolisme du fer : la plupart sont des maladies à transmission autosomique récessive mais certaines sont dominantes en particulier l’hémochromatose HFE4 ; la plupart correspondent à des accumulations cytosoliques de fer sous forme de ferritine, mais certaines, en particulier l’ataxie de Friedreich sont des anomalies du métabolisme mitochondrial avec accumulation du métal dans ces organites subcellulaires.
Summary
The classification of hereditary abnormalities of iron metabolism was recently expanded and diversified. Genetic hemochromatosis now corresponds to six diseases, namely classical hemochromatosis HFE 1 ; juvenile hemochromatosis HFE 2 due to mutations in an unidentified gene on chromosome 1 ; hemochromatosis HFE 3 due to mutations in the transferrin receptor 2 (TfR2) ; hemochromatosis HFE 4 caused by a mutation in the H subunit of ferritin ; and hemochromatosis HFE 6 whose gene is hepcidine (HAMP). Systemic iron overload is also associated with aceruloplasminemia, atransferrinemia and the ‘‘ Gracile ’’ syndrome caused by mutations in BCS1L. The genes responsible for neonatal and African forms of iron overload are unknown. Other genetic diseases are due to localized iron overload : Friedreich’s ataxia results from the expansion of triple nucleotide repeats within the frataxin (FRDA) gene ; two forms of X-linked sideroblastic anemia are due to mutations within the δ aminolevulinate synthetase (ALAS 2) or ABC-7 genes ; Hallervorden-Spatz syndrome is caused by a pantothenate kinase 2 gene (PANK-2) defect ; neuroferritinopathies ; and hyperferritinemia— cataract syndrome due to a mutation within the L-ferritin gene. In addition to this wide range of genetic abnormalities, two other features characterize these iron disorders : 1) most are transmitted by an autosomal recessive mechanism, but some, including hemochromatosis type 4, have dominant transmission ; and 2) most correspond to cytosolic iron accumulation while some, like Friedreich’s ataxia, are disorders of mitochondrial metabolism.
Les anomalies du métabolisme du fer sont connues depuis la fin du 19ème siècle, puisque l’hémochromatose d’abord décrite sous le terme de cirrhose bronzée par Trousseau en 1865, a ensuite été rattachée à une surcharge tissulaire martiale par Recklinghausen en 1889. La survenue tardive de l’affection, la diversité et la relative non spécificité de la symptomatologie clinique, expliquent qu’il ait fallu attendre le milieu des années 1970 pour que soient démontrés son caractère héréditaire et sa transmission autosomique récessive [1] [2]. L’hémochromatose a pourtant été l’une des rares maladies autosomiques dont le gène ait bénéficié, bien avant le développement de la génétique moléculaire, d’une assignation chromosomique (en 6p21) et ceci grâce à une association forte avec l’antigène sérologique HLA-A3. Le gène, maintenant appelé HFE 1, a été identifié en 1996 [3]. Ce clonage a conduit au démembrement nosologique de « l’hémochromatose génétique » ; il a également coïncidé avec un large accroissement des anomalies héréditaires du métabolisme du fer, parmi lesquelles s’individualise aujourd’hui un groupe de mitochondriopathies.
LES HÉMOCHROMATOSES GÉNÉTIQUES
Hémochromatose type 1 (HFE 1)
La maladie est due a une dysrégulation de l’absorption intestinale du fer, entraînant une lente et progressive accumulation de ce métal dans les tissus, en particulier dans le foie, avec prédominance hépatocytaire et péricentrolobulaire. La traduction biologique est l’augmentation de la sidérémie, du pourcentage de saturation de la transferrine, de la ferritinémie et de la concentration tissulaire hépatique en fer.
Avant que ne soit pratiqué un traitement précoce par saignée, la conséquence de ces perturbations biologiques était le développement d’une symptomatologie variée, habituellement entre 35 et 45 ans chez l’homme, plus tardivement chez la femme pour des raisons physiologiques (pertes de fer du fait des règles et des grossesses) :
mélanodermie, cirrhose évoluant dans un tiers des cas vers l’hépatocarcinome, diabète insulino-dépendant, insuffisance hypophysogonadique, cardiomyopathie hypertrophique, arthropathies dont la plus caractéristique, bien que non pathognomonique, est une arthrite chronique des 2ème et 3ème articulations métacarpophalangiennes.
Les résultats du traitement, représenté par des saignées régulières, dépendaient étroitement de la précocité du diagnostic : au stade d’hémochromatose cliniquement confirmée, seule la cardiomyopathie était régressive alors que la plupart des autres atteintes tissulaires (cirrhose, diabète, arthropathie, endocrinopathies) ne l’étaient pas [4]. Ainsi s’est rapidement justifiée l’idée d’un traitement précoce, mis en œuvre avant l’apparition des signes cliniques et reposant donc sur le diagnostic biologique de l’affection. Une telle stratégie a fait rapidement disparaître le tableau classique de la maladie qui ne s’observe plus que rarement voire exceptionnellement. Le diagnostic biologique n’est cependant pas spécifique, il est parfois conforté par des signes mineurs tels qu’asthénie, arthralgie, augmentation inexpliquée de transaminases. Il est le plus souvent porté de façon fortuite à l’occasion d’un bilan biologique ou dans le cadre d’une consultation génétique dans une famille de proband. Dans ces conditions, l’apport de la génétique moléculaire, c’est-à-dire l’analyse du gène HFE 1, est devenu déterminant.
Le Gène HFE 1
Le gène HFE 1, cloné en 1996, est situé en 6p22 à environ 4,5 mégabases télomériques à HLA-A. Il s’agit d’un gène d’environ 12 kb comprenant 7 exons et codant pour une protéine présentant de très fortes homologies avec les molécules de classe I du complexe majeur d’histocompatibilité. Cette protéine est en effet une glycoprotéine de 343 acides aminés comportant un domaine transmembranaire, une extré- mité COOH terminale intracytoplasmique et trois domaines extra cellulaires α , 1 α2 et α ; le domaine « immunoglobulin like » se lie à la microglobuline, et dans le 3 α3 β2 domaine α un groupe de 4 résidus histidine représente potentiellement un site de 1
liaison de HFE à une autre protéine. Malgré les très fortes similitudes avec les molécules HLA classe I, en particulier HLA A et HLA G, les études structurales 2 montrent que HFE n’est capable ni de lier les peptides endogènes, ni de fixer les métaux avec une grande affinité.
Lors de la découverte de HFE, l’implication d’un gène HLA classe I dans le métabolisme du fer était bien entendu loin d’être évidente. Un élément en ce sens avait pourtant été l’observation d’une surcharge en fer chez les souris invalidées pour le gène de la β microglobuline. [5]. Un autre argument était la très forte 2 proportion d’une mutation faux-sens (C282Y) à l’état homozygote chez les malades.
La preuve définitive a été apportée par l’étude des souris HFE-/- (knock-out, absence complète de la protéine) et des souris transgéniques C282Y +/+ (knock-in, présence de la protéine mutée) qui présentent un phénotype de surcharge en fer semblable à celui de l’hémochromatose [6] [7].
En absence de voie spécifique d’excrétion, l’homeostasie du fer repose sur une régulation étroite de la voie intestinale d’absorption, essentiellement duodénale.
Cette régulation serait sous le contrôle de deux messagers hormonaux, l’un modulé par l’état des réserves tissulaires en fer (il s’agirait potentiellement de l’hepcidine), l’autre par les besoins de l’erythropoièse. Le rôle de HFE dans cette régulation de l’absorption intestinale du fer est démontré par la physiopathologie de l’hemochromatose, mais les mécanismes biochimiques en cause restent spéculatifs ; la seule notion établie est celle d’une interaction, à la membrane plasmique de divers types cellulaires, entre HFE et le récepteur de la transferrine diminuant dans un rapport de 5 à 10 l’affinité de celui-ci pour son ligand et entraînant donc une diminution de la captation du fer [8]. Dans une première hypothèse, la protéine HFE participerait dans les cellules des cryptes intestinales à la programmation des enterocytes diffé- rentiés en vue d’adapter leurs capacités d’absorption du fer au besoin de l’organisme. Il a été ensuite proposé que HFE régulerait l’absorption ou le relarguage du fer à partir des cellules du système reticuloendothélial et des cellules intestinales, en fonction du pourcentage de saturation de la transferrine et des concentrations de son récepteur dans le sérum [9]. Enfin récemment l’hypothèse a été émise que le point central de cette régulation serait hépatique et non plus duodénale : les hépatocytes, sensibles à l’état du capital ferrique dans les complexes ternaires HFE/recepteur de transferrine/transferrine, y réagiraient par une sécrétion appropriée d’hepcidine, qui à son tour modulerait dans les entérocytes l’expression de Ireg1, c’est-à-dire du canal transporteur de fer au niveau de leur membrane basolatérale [10].
Mutations de HFE
C282Y
Située dans l’exon 4, elle correspond a une transition guanine/adénine avec pour conséquence le remplacement de la cystéine 282 par une tyrosine. La mutation
C282Y entraîne la disparition du pont disulfure conditionnant la configuration « immunoglobulin like » du domaine α , le changement de structure spatiale de la 3 molécule empêchant son association à la β microglobuline. La mutation est pré- 2 sente à l’état homozygote chez plus de 95 % des sujets diagnostiqués comme hémochromatosiques dans le Nord-Ouest de l’Europe (Bretagne, Irlande…) [11] et seule cette homozygotie est considérée aujourd’hui comme responsable de l’hémochromatose HFE 1, maladie à transmission autosomique récessive. Chez les patients, il existe un gradient décroissant Nord-Ouest/Sud-Est de cette homozygotie C282Y, les fréquences les plus faibles étant observées en Italie (64 %) [12] et en Grèce (30 %) [13]. Ces données ont très vite suggéré l’existence d’autres formes d’hémochromatoses, en relation avec les origines ethniques des populations concernées.
La mutation C282Y présente un même gradient de fréquence Nord-ouest/Sud-Est dans la population européenne [14], les fréquences les plus élevées d’hétérozygotes s’observant en Irlande (20 %) et en Bretagne (16 %). Elle est par contre absente dans les populations Africaines ou asiatiques. Elle apparaît donc spécifique des populations européennes ou d’origine européennes (Etats-Unis, Australie)…. La mutation C282Y est dans la majorité des cas retrouvée sur un chromosome 6 à configuration allélique particulière, qualifiée « d’haplotype ancestral », sur une distance d’environ 6 mégabases englobant HLA-A et s’étendant vers le télomére. Cette caractéristique suggère pour les généticiens des populations que la mutation serait d’apparition récente (60 à 100 générations, c’est-à-dire environ 2000 ans). Apparue dans le Nord de l’Europe, sa diffusion avec une telle rapidité ne peut être que le résultat d’un avantage sélectif important conféré aux porteurs de la mutation, par exemple une plus grande résistance aux nombreuses pandémies qui ont ravagé l’Europe, en particulier au moyen-âge.
Dans le Nord de l’Europe la fréquence des sujets homozygotes C282Y est donc d’environ 5 à 6 p. 1000 [14] [15]. L’hémochromatose HFE 1 apparaîtrait ainsi comme la plus fréquente des maladies héréditaires et ses caractéristiques, en particulier l’existence d’un traitement simple, efficace et peu coûteux, justifieraient a priori un dépistage systématique. Cette idée est depuis peu contrebalancée par un autre concept : celui de la faible pénétrance de l’homozygotie C282Y. Les données en faveur de cette faible expressivité se sont progressivement accumulées : disproportion à Jersey entre le petit nombre de cas d’hémochromatose inscrits au registre consacré à cette maladie et la fréquence élevée de la mutation dans la population générale [16] ; fréquence d’homozygotes C282Y aussi élevée chez les personnes âgées se présentant aux urgences de l’hôpital de Norwich (UK) pour divers motifs, que dans la population générale [17]. Plusieurs études récentes de population en Australie, au Canada, en Nouvelle Zélande, aux Etats Unis, en Angleterre [18], en Bretagne [19] aboutissent à des conclusions semblables : l’expression clinique est peu fréquente, certains auteurs évoquent même une pénétrance d’environ 1 % [20] ; par ailleurs l’expression biologique retrouvée chez tous les hommes, n’est observée que chez environ la moitié des femmes [18]. Les facteurs, en particulier génétiques, qui conditionnent cette variabilité d’expression clinique et biologique de l’hémochro-
matose sont encore inconnus [21] [22]. L’hypothèse d’un digénisme, c’est-à-dire de mutations concomittantes dans deux gènes différents, a été faite : l’observation de quelques malades homozygotes C282Y présentant des mutations du gène de l’hepcidine va dans ce sens [23]. Quoiqu’il en soit, la faible pénétrance clinique de la maladie est bien entendu l’un des paramètres à prendre en compte dans les réflexions qui sont régulièrement menées sur l’intérêt et les modalités d’un dépistage systématique de l’affection.
Autres mutations
La publication originale de Feder et al [3] identifiait une deuxième mutation H63D, remplacement d’une histidine par un acide aspartique dans le domaine extracellulaire α . Cette mutation faux-sens a une distribution mondiale, avec une fréquence 1 d’environ 20 % dans les populations caucasoïdes [24]. Une littérature abondante a été consacrée au rôle pathogène éventuel de cette mutation ; rien de déterminant ne s’en dégage. Tout au plus doit-on considérer H63D comme un polymorphisme ayant dans un contexte génétique particulier, tel que l’association avec C282Y, un faible rôle favorisant la surcharge en fer. De telles surcharges martiales n’appartiennent pas au cadre de l’hémochromatose génétique, au sens mendélien du terme ; il s’agit d’un vaste groupe d’affections multifactorielles dont l’inventaire des déterminants génétiques et environnementaux est loin d’être achevé. Les mêmes réflexions peuvent également concerner la mutation S65C d’abord décrite comme un simple polymorphisme [25], puis comme facteur favorisant la surcharge [26].
Une dizaine d’autres mutations non sens, faux-sens ou introniques entraînant un défaut d’épissage de l’ARN messager ont été décrites [27]. Elles sont très rares, voire privées.
Hémochromatose type 2A (HFE 2A) ou hémochromatose juvénile
L’hémochromatose juvénile est une affection rare, à transmission autosomique récessive, dont le tableau clinique s’individualise simplement par une évolution accélérée dès la deuxième ou la troisième décade de la vie, avec au premier plan cardiomyopathie et hypogonadisme. Le gène a été localisé en 1999 en 1q21, dans une région riche en gènes mais dont aucun n’évoque un lien particulier avec le métabolisme du fer tel qu’il est connu à ce jour [28]. Etant donné la rapidité d’installation du tableau clinique, le gène HFE 2 joue vraisemblablement un rôle déterminant dans la régulation de l’absorption intestinale du fer.
Hémochromatose type 3 (HFE 3)
L’hémochromatose type 3 est également une maladie à transmission autosomique récessive, dont le tableau clinico-biologique est semblable à celui de HFE 1. Elle est due à des mutations affectant le deuxième récepteur de la transferrine (TfR ). Ce 2
gène, localisé en 7q22, étendu sur environ 20 kb, formé de 18 exons, donne naissance par épissage alternatif à deux isoformes principales dépourvues de motif IRE (« Iron Responsive Element ») : un transcrit α englobant la totalité de la séquence codante et un transcrit β dépourvu des trois premiers exons [29]. La forme α est exprimée à un taux élevé dans la lignée érythroïde et dans les hépatocytes ; la forme β l’est à taux faible mais de façon ubiquitaire. La protéine α est transmembranaire et possède un large domaine extra-cytoplasmique homologue au premier récepteur de la transferrine (TfR1), la protéine β dépourvue de domaine transmembranaire est intracellulaire.
Le rôle de TfR2 dans le métabolisme du fer est encore loin d’être compris, la fixation de transferrine se faisant avec une affinité plus faible que dans le cas du premier récepteur (TfR1). Par ailleurs, l’absence de motif IRE semble indiquer que TfR2 échappe au système de régulation post-transcriptionnelle intra cellulaire du métabolisme du fer par les « Iron Responsive Protein » (IRP). Le meilleur argument pour souligner l’importance de TfR2 dans le métabolisme du fer est comme très souvent la pathologie et la description de mutations ségrégeant avec des surcharge martiales dans des familles consanguines. Quatre mutations différentes ont été décrites à l’état homozygote dans 5 familles de Sicile et d’Italie [30] [31] [32].
Hémochromatose type 4 (HFE 4)
L’hémochromatose type 4 est provoquée par des anomalies du gène d’un transporteur trans-membranaire du fer : SLC11A3 communément appelé ferroportine 1 (FPM1) [33]. Ce gène identifié assez paradoxalement comme responsable chez le poisson Zebrafish d’une variété d’anémie génétique (Weissherbst) s’étend sur environ 20 kb en 2q32 ; constitué de 8 exons, il code une protéine comportant 9 ou 10 domaines trans-membranaires selon les bases de données [34]. Chez l’Homme, il est exprimé fortement dans le syncytiotrophoblaste puis le placenta, les macrophages, les cellules de Küpfer, les hépatocytes, les reins et les entérocytes dans lesquels la protéine a une localisation dans la membrane baso-latérale. L’ARN messager correspondant contient un motif IRE dans sa partie 5′ UTR ce qui est donc en faveur d’un mécanisme de régulation post-transcriptionnel par le taux de fer intracellulaire [35].
Le tableau provoqué par des mutations de la ferroportine présente trois particularités essentielles par rapport aux autres hémochromatoses : la biopsie hépatique caractérise une accumulation de fer largement prédominante dans les cellules réticulo-endothéliales, la transmission de l’affection est autosomale dominante et la sémiologie biochimique est marquée par une discordance entre une hyperferritiné- mie et une saturation de la transferrine normale ou subnormale. Une autre caractéristique de l’affection mérite également d’être soulignée : elle apparaît à ce jour comme la plus fréquente des hémochromatoses autres que HFE 1. Identifiée dans un premier temps dans une famille hollandaise [36], HFE 4 a été ensuite rapidement décrite en Australie, Italie, Angleterre, Grèce, France…. L’une des mutations, la
délétion V162del est retrouvée dans différents sites géographiques, ce qui suggère une multiplicité des évènements mutationnels. L’essentiel serait cependant de comprendre la physiopathologie de la surcharge martiale : gain de fonction, perte de fonction, haplo-insuffisance ont été invoqués, mais les mécanismes biochimiques en cause restent très largement inconnus.
Hémochromatose type 5 (HFE 5) : H ferritine
Il s’agit d’un syndrome de surcharge martiale, décrit dans une famille japonaise [37], dont l’expression phénotypique est sensiblement identique à celle de l’hémochromatose classique HFE1, mais dont le caractère particulier est une transmission autosomique dominante. Ce phénotype ségrège avec une conversion A → U, à l’état hétérozygote, retrouvée en position 49 de l’ARN messager de la sous-unité H-ferritine, c’est-à-dire dans la boucle IRE. Les mécanismes biochimiques en cause dans l’accumulation de fer sont pour le moment mal compris.
Hémochromatose type 2B (HFE 2B) : Hepcidine (HAMP)
L’hepcidine est apparue récemment comme un acteur clef du métabolisme du fer, potentiellement comme le régulateur humoral des stocks de fer dont l’existence est postulée depuis fort longtemps. D’abord caractérisée pour son activité antibactérienne et isolée du sang et de l’urine, l’hepcidine a été rattachée à la régulation du métabolisme martial sur les constats successifs de son augmentation d’expression hépatique au cours des surcharges expérimentales [38], de l’accumulation hépatique en fer chez les souris dont le gène HAMP était inhibé [39] et enfin de l’apparition d’une anémie ferriprive chez les souris transgéniques surexprimant ce gène [40].
Ce peptide de 25 acides aminés, dans sa forme mature la plus importante, est riche en cystéine et présente une configuration spatiale avec quatre ponts disulfure. Le gène (HAMP) est localisé en 19q13 et comprend 3 exons. Récemment deux mutations de ce gène ont été rapportées dans deux familles grecques consanguines [41] : une délétion d’un nucléotides (93delG) dans l’exon 2 entraînant la biosynthèse d’un propeptide anormal et allongé ; une mutation stop (R56X) dans l’exon 3. Dans ces deux familles il s’agissait d’un tableau qualifié d’hémochromatose juvénile ; ce phénotype recouvre donc plusieurs entités génétiques différentes.
AUTRES FORMES DE SURCHARGES MARTIALES GÉNÉRALISÉES
Hémochromatose néo-natale :
L’hémochromatose néo-natale correspond à une surcharge martiale massive, en particulier hépatique, constituée in utero et très rapidement mortelle. La seule
possibilité thérapeutique est la transplantation hépatique. Il est vraisemblable que l’affection correspond à un syndrome aux étiologies multiples tant somatiques telles que des infections virales, que génétiques. Plusieurs modes de transmission ont été proposés : autosomique récessif, transmission maternelle évoquant un mosaïcisme ou une mitochondriopathie [42]. Aucune anomalie des gènes connus du métabolisme du fer n’a été retrouvée à ce jour.
Surcharge ferrique des Africains
Les surcharges ferriques sont fréquentes en Afrique sub-saharienne, où elles entraî- nent fibrose portale hépatique et cirrhose, souvent compliquée d’hépatocarcinome.
Elle sont considérées également comme un facteur favorisant différentes maladies infectieuses, en particulier la tuberculose. L’examen histologique montre chez les malades, une accumulation de fer touchant à la fois les hépatocytes et les cellules du système réticulo-endothélial. D’abord considérées comme exclusivement d’origine alimentaire en raison de la consommation de breuvages fermentés riches en fer, ces surcharges ont été ensuite également rattachées à un facteur génétique non lié au chromosome 6, c’est-à-dire à HFE1. L’existence d’un tel facteur génétique est également démontrée chez les Afro-Américains atteints de surcharge martiale primitive. Aucune mutation de HFE1, de TfR2 ou de la ferroportine n’a été retrouvée chez ces malades [43].
Acéruloplasminémie
L’acéruloplasminémie est une rare affection autosomale récessive, surtout observée dans la population japonaise. Diverses mutations du gène de la céruloplasmine, localisé en 3q21-24, ont été décrites ; elles font disparaître son activité oxydase entraînant un défaut de recyclage du fer provenant pour l’essentiel du catabolisme de l’hémoglobine [44]. Les conséquences en sont une insuffisance de l’érythropoïèse et une accumulation tissulaire de fer, y compris dans le système nerveux central. Le tableau clinique est riche : anémie, insuffisance hépatique avec dépôt de fer aussi bien dans les hépatocytes que dans les cellules du système réticulo-endothélial, diabète, dégénérescence rétinienne, ataxie cérébelleuse et démence.
Atransferrinémie
L’atransferrinémie est extrêmement rare puisque seuls 10 cas dans 8 familles ont été rapportés ; plusieurs de ces sujets sont morts très jeunes, les autres ont été maintenus en vie par des transfusions de plasma ou de transferrine purifiée. Son absorption intestinale est augmentée, mais le fer est difficilement utilisable pour l’érythropoïèse, avec pour conséquence une anémie hypochrome microcytaire sévère. Par ailleurs, une accumulation de fer se constitue progressivement dans le foie, le pancréas, les reins, le myocarde et la glande thyroïde.
Syndrome « Gracile »
Il s’agit d’une affection autosomale récessive, diagnostiquée chez 25 enfants appartenant à 18 familles en majorité finlandaise. L’essentiel du tableau est regroupé sous le terme de « Gracile », acronyme de « Growth retardation, amino-aciduria type Fanconi, cholestasis, iron overload, lactacidosis and early death » [46] ; à ce syndrome clinique, il convient d’ajouter surcharge martiale hépatique, hypersidérémie, augmentation de la saturation de la transferrine. Le gène responsable de ce syndrome est BCS1L ; il code pour une protéine de la membrane interne des mitochondries, connue pour être une protéine chaperonne nécessaire au bon assemblage du complexe III de la chaîne respiratoire. Une mutation unique (S78G) est retrouvée à l’état homozygote chez l’ensemble des malades finlandais, atteints du syndrome Gracile mais ces malades ne présentent pas de déficit d’activité du complexe III.
Récemment, cinq mutations de ce gène ont été rapportées chez 3 patients anglais, ainsi que quatre mutations chez des malades turcs se présentant avec un tableau de déficit de la chaîne respiratoire mitochondriale [47]. Les relations entre BCS1L et métabolisme du fer sont à ce jour totalement inconnues.
SURCHARGES LOCALISÉES
Ataxie de Friedreich
L’ataxie de Friedreich est une maladie à transmission autosomique récessive. Le gène localisé en 9q13 appartient donc à l’ADN nucléaire, mais code pour une protéine mitochondriale, la frataxine [48]. Dans l’immense majorité des cas, l’anomalie est une expansion du triplet GAA dans l’intron 1 du gène, entraînant une réduction de sa transcription et de sa traduction. La diminution de la concentration en frataxine a pour conséquence l’accumulation de fer dans les mitochondries, entraînant une augmentation des réactions de stress oxydatif et expliquant l’atteinte préférentielle de cellules spécifiquement sensibles comme les cardiomyocytes et certains neurones.
Le rôle de la frataxine dans le métabolisme du fer mitochondrial n’est pas entièrement compris. Le travail expérimental est cependant facilité par l’existence d’un modèle animal particulièrement simple puisqu’il s’agit de Saccharomyces cerevisiae : l’orthologue de la frataxine chez la levure est appelé YFH 1 ; il existe des mutants déficitaires qui présentent également une accumulation de fer dans les mitochondries. Dans ce modèle, la frataxine interviendrait dans l’incorporation du fer dans les protéines à groupement fer-soufre [49] et dans l’utilisation du fer par la ferrochélatase [50].
Anémies sidéroblastiques liées à l’X
Il y a deux formes d’anémies sidéroblastiques liées à l’X, l’une avec et l’autre sans ataxie. Les deux formes, s’accompagnent d’une diminution de l’efficacité de la biosynthèse de l’hème, conduisant à une accumulation de fer dans la mitochondrie.
La forme sans ataxie est due à des mutations de la δ amino-levulinate synthétase (ALAS2), premier système enzymatique et enzyme clef de la voie de biosynthèse des porphyrines et de l’hème, et dont le gène est localisé en X q11-21. Par ailleurs un certain nombre de patients répondent à des doses pharmacologiques de pyridoxine, co-facteur de l’ALAS2.
L’anémie sidéroblastique avec ataxie correspond à des anomalies de ABC-7, protéine appartenant à la grande famille des protéines ABC, liant l’ATP et dont le gène est localisé en Xq13.1-q13.3. Le lien entre ABC-7, métabolisme du fer et biosynthèse de l’hème est encore mal compris ; il a été montré que chez Saccharomyces Cerevisiae, la protéine orthologue (ATm 1p) était impliquée dans le transport des groupements « fer-soufre » depuis leur site de biosynthèse dans la mitochondrie vers le cytoplasme. ABC-7 régulerait l’expression de la ferrochelatase, également protéine à cluster fer-soufre [51].
Syndrome d’Hallervorden-Spatz
Le syndrome d’Hallervorden-Spatz, décrit en 1922, est une affection neurodégéné- rative, à transmission autosomale récessive, associée à une accumulation de fer dans le globus pallidus et la pars reticula de la substantia nigra. La maladie débute au cours de la première ou deuxième décade de la vie et le tableau confirmé associe un syndrome extra pyramidal avec dystonie, rigidité, choréoathétose, une rétinite pigmentaire avec atrophie optique, un syndrome parkinsonien et une démence progressive. Par clonage positionnel, le gène a été localisé en 20p12.3 et des mutations du gène de la pantothénate kinase 2 (PANK2) ont été identifiées [52]. Le gène est transcrit et traduit en deux isoformes dont l’une serait spécifiquement mitochondriale, et responsable de l’affection [53]. La pantothénate-kinase catalyse la première des cinq étapes de biosynthèse du coenzyme A ; le phosphopantothénate obtenu se condense normalement avec la cystéine. En cas de déficit d’activité de la pantothé- nate kinase, il y a donc accumulation de cystéine dont les propriétés de chélation du fer seraient responsables de l’accumulation du métal dans les mitochondries.
Neuroferritinopathies
Décrite en 2001, chez des patients de 40 à 55 ans originaires du Nord de l’Angleterre, cette affection est caractérisée par des mouvements involontaires et un syndrome extrapyramidal, mais sans atteinte des fonctions cognitives. Tous les malades étaient hétérozygotes pour l’insertion d’une adénine en position 460 dans le gène de la L-ferritine [54]. La conséquence de cette insertion est un décalage du cadre de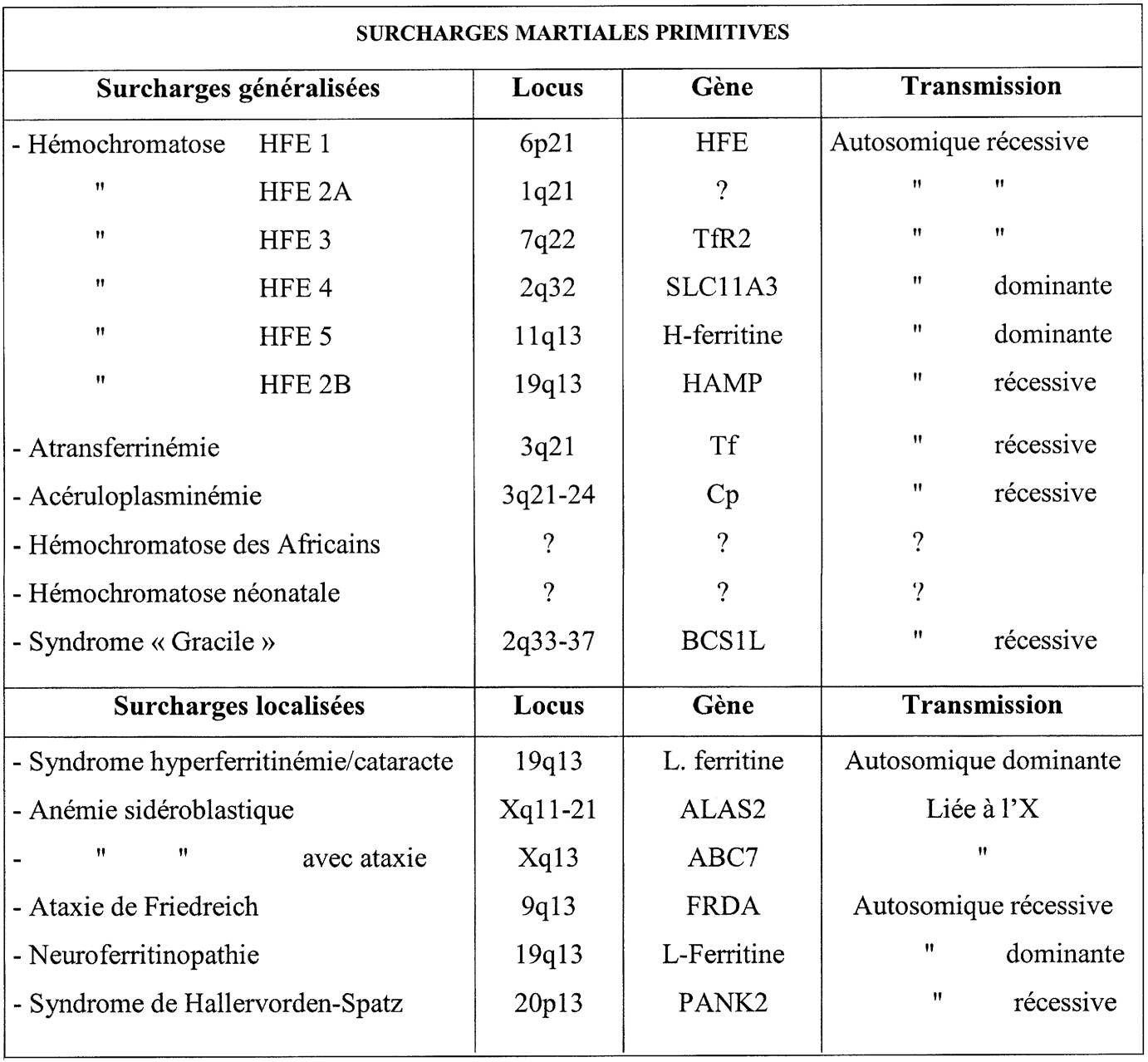 lecture dans la partie C terminale du gène. L’examen histologique du cerveau montre une accumulation importante de fer et de ferritine dans les neurones du globus pallidus, mais aussi dans le cerveau antérieur et le cervelet. Ces patients n’ont par ailleurs aucune anomalie sérique du métabolisme du fer, avec en particulier une ferritinémie normale voire basse.
lecture dans la partie C terminale du gène. L’examen histologique du cerveau montre une accumulation importante de fer et de ferritine dans les neurones du globus pallidus, mais aussi dans le cerveau antérieur et le cervelet. Ces patients n’ont par ailleurs aucune anomalie sérique du métabolisme du fer, avec en particulier une ferritinémie normale voire basse.
Syndrome hyperferritinémie-cataracte
Le syndrome hyperferritinémie-cataracte, à transmission dominante, n’est pas en réalité une anomalie du métabolisme du fer : la sidérémie est normale et la ponction hépatique ne montre aucune anomalie de surcharge. Il touche néanmoins une protéine de ce métabolisme puisqu’il est provoqué par des mutations dans le motif IRE du gène de la L Ferritine [55]. Une dizaine de mutations a été décrite dans ce motif IRE ; elles ont pour conséquence de réduire l’affinité de l’IRE de l’ARN messager de la ferritine pour les IRP (« Iron Responsive Protein »), entraînant donc une traduction de l’ARN messager non régulée par les concentrations intracellulai-
res en fer. La cataracte est due à des dépôts de ferritine, sous forme de cristaux, à l’intérieur du cristallin.
Les surcharges martiales primitives, dont le nombre est sans doute encore appelé à augmenter, apportent donc des éclairages nouveaux sur le métabolisme du fer. Au delà de la diversité d’expression phénotypique de ces maladies, deux notions méritent ainsi d’être soulignées : celle d’anomalies du métabolisme mitochondrial et celle d’anomalies touchant spécifiquement le tissu cérébral. Dans l’un et l’autre cas, les voies métaboliques en cause restent très largement à éclaircir.
BIBLIOGRAPHIE [1] SIMON M., BOUREL M., FAUCHET R., GENETET B. — Association of HLA-A3 antigens with idiopathic haemochromatosis . GUT , 1976, 1976 , 332-334.
[2] SIMON M., BOUREL M., GENETET B., FAUCHET B. Idiopathic hemochromatosis. — Demonstration of recessive transmission and early detection by family HLA typing. N. Engl. J. Med ., 1977, 297 , 1017-1021.
[3] FEDER J.N., GNIRKE A., THOMAS W., TSUCHIHASHI Z., RUDDY D.A., BASAVA A. et al . — A novel
MHC class I-like gene is mutated in patients with hereditary haemochromatosis.
Nat. Genet ., 1996, 13 , 399-408.
[4] NIEDEREAU C., FISCHER R., SONNENBERG A., STREMMEL W., TRAMPISH H.J., STROMEYER G. — Survival and causes of death in cirrhotic and non-cirrhotic patients with primary hemochromatosis. N. Engl. J. Med., 1985, 14 , 1256-1262.
[5] DE SOUSA M., REIMAO R., LACERDA R., HUGO P., KAUFMANN S.H., PORTO G. — Iron overload in beta 2-microglobulin-deficient mice. Immunol . Lett ., 1994, 39 , 105-111.
[6] ZHOU X.Y., TOMATSU S., FLEMING R.E., PARKKILA S., WAHEED A., JIANG J. et al. — HFE gene knockout produces mouse model of hereditary hemochromatosis.
Proc. Natl. Acad. Sci. U S A., 1998, 95 , 2492-2497.
[7] LEVY J.E., MONTROSS L.K., COHEN D.E., FLEMING M.D., ANDREWS N.C — Hereditary hemochromatosis does not produce a null allele. Blood , 1999, 94 , 9-11.
[8] ROY C.N., PENNY D.M., FEDER J.N., ENNS C.A. — The hereditary hemochromatosis protein, HFE, specifically regulates transferrin-mediated iron uptake in HeLa cells. J. Biol. Chem., 1999, 274 , 9022-9028.
[9] TOWNSEND A., DRAKESMITH H. — Role of HFE in iron metabolism, hereditary haemochromatosis, anaemia of chronic disease, and secondary iron overload. The Lancet , 2002, 359 , 786-790.
[10] FRAZER D.M., ANDERSON G.J. — The orchestration of body iron intake : how and where do enterocytes receive their cues ? Blood Cells, Molecules, and Diseases , 2003, 30 , 288-297.
[11] JOUANOLLE A.M., GANDON G., JEZEQUEL P., BLAYAU M., CAMPION M.L., YAOUANQ J. et al. —
Haemochromatosis and HLA-H.
Nat. Genet ., 1996, 14 , 251-252.
[12] CARELLA M., D’AMBROSIO L., TOTARO A., GRIFA A., VALENTINO M.A., PIPERNO A. et al. —
Mutation analysis of the HLA-H gene in Italian hemochromatosis patients.
Am. J. Hum.
Genet., 1997, 60 , 828-832.
[13] PAPANIKOLAOU G., POLITOU M., TERPOS E., FOURLEMADIS S., SAKELLAROPOULOS N., LOUKOPOULOS D. — Hereditary hemochromatosis : HFE mutation analysis in Greeks reveals genetic heterogeneity. Blood Cells Mol. Dis., 2000, 26 , 163-168.
[14] ALISON T., MERRYWEATHER-CLARKE A.T., POINTON J.J., JOUANOLLE A.M., ROCHETTE J. et al. —
Geography of HFE C282Y and H63D mutations.
Genet. Test ., 2000, 4 (2), 183- [15] JOUANOLLE A.M., FERGELOT P., RAOUL M.L., GANDON G., ROUSSEY M., DEUGNIER Y. et al. —
Prevalence of the C282Y mutation in Brittany : penetrance of genetic hemochromatosis ?
Ann.
Genet ., 1998, 41 , 195-198 [16] MERRYWEATHER-CLARKE A.T., WORWOOD M., PARKINSON L., MATTOCK C., POINTON J.J., ShEARMAN J.D. et al. — The effect of HFE mutations on serum ferritin and transferrin saturation in the Jersey population.
Br. J. Haematol ., 1998, 101 , 369-373.
[17] WILLIS G., WIMPERIS J.Z., SMITH K.C., FELLOWS I.W., JENNINGS B.A. — Haemochromatosis gene C282Y homozygotes in an elderly male population. Lancet , 1999, 354 , 221-222.
[18] MCCUNE C.A., AL-JADER L.N., MAY A., HAYES S.L., JACKSON H.A., WORWOOD M. — Hereditary haemochromatosis : only 1 % of adult HFEC282Y homozygotes in South Wales have a clinical diagnosis of iron overload. Hum Genet ., 2002, 111 , 538-543.
[19] DEUGNIER Y., JOUANOLLE A.M., CHAPERON J., MOIRAND R., PITHOIS C., MEYER J.F. et al. —
Gender-specific phenotypic expression and screening strategies in C282Y— linked haemochromatosis : a study of 9396 French people. Br. J. Haematol ., 2002, 118 , 1170-1178.
[20] BEUTLER E., FELITTI V.J., KOZIOL J.A., HO N.J., GELBART T. — Penetrance of 845G-> A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet , 2002, 359 , 211-218.
[21] SACHOT S., MOIRAND R., JOUANOLLE A.M., MOSSER J., FERGELOT P., DEUGNIER Y. et al. — Low penetrant hemochromatosis phenotype in eight families : no evidence of modifiers in the MHC region. Blood Cells Mol. Dis., 2001, 27 , 518-529.
[22] BEUTLER E. — The HFE Cys282Tyr mutation as a necessary but not sufficient cause of clinical hereditary hemochromatosis. Blood , 2003, 101 , 3347-3350.
[23] MERRYWEATHER-CLARKE A.T., CADET E., BOMFORD A., CAPRON D., VIPRAKASIT V. et al . —
Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet ., 2003, 17 , 2241-2247.
[24] HANSON E.H., IMPERATORE G. & BURKE W. — HFE gene and hemochromatosis : a HuGE review. Am. J. Epidemiol ., 2001, 154 , 193-206 [25] DOUABIN V., MOIRAND R., JOUANOLLE A., BRISSOT P., LE GALL J.Y., DEUGNIER Y. et al. —
Polymorphisms in the HFE gene.
Hum. Hered ., 1999, 49 , 21-26.
[26] MURA C., RAGUENES O., FEREC C. HFE mutations analysis in 711 hemochromatosis probands :
evidence for S65C implication in mild form of hemochromatosis . Blood, 1999 , 93, 2502-2505.
[27] POINTON J.J., WALLACE D., MERRYWEATHER-CLARKE A.T., ROBSON K.J. — Uncommon mutations and polymorphisms in the hemochromatosis gene. Genet. Test ., 2000, 4 , 151-161 [28] ROETTO A., TOTARO A., CAZZOLA M., CICILANO M., BOSIO S., D’ASCOLA G. et al. — Juvenile hemochromatosis locus maps to chromosome 1q.
Am. J. Hum. Genet . 1999, 64 , 1388-1393.
[29] KAWABATA H., YANG R., HIRAMA T., VUONG P.T., KAWANO S., GOMBART A.F. et al. —
Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol. Chem ., 1999, 274 , 20826-20832.
[30] CAMASCHELLA C., ROETTO A., CALI A., De GOBBI M., GAROZZO G., CARELLA M. et al. — The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22.
Nat Genet ., 2000, 25 , 14-15.
[31] ROETTO A., TOTARO A., PIPERNO A., PIGA A., LONGO F., GAROZZO G. et al. — New mutations inactivating transferrin receptor 2 in hemochromatosis type 3.
Blood , 2001, 97 , 2555-2560.
[32] GIRELLI D., BOZZINI C., ROETTO A., ALBERTI F., DARAIO F., COLOMBARI R. et al. — Clinical and pathologic findings in hemochromatosis type 3 due to a novel mutation in transferrin receptor 2 gene. Gastroenterology , 2002, 122 , 1295-1302.
[33] ABBOUD S., HAILE D.J. — A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem ., 2000, 275 , 19906-19912.
[34] DONOVAN A., BROWNLIE A., ZHOU Y., SHEPARD J., PRATT S.J., MOYNIHAN J. et al . — Positional cloning of zebrafish ferroportin identifies a conserved vertebrate iron exporter.
Nature , 2000, 403 , 776-781.
[35] EISENSTEIN R.S. — Iron regulatory proteins and the molecular control of mammalian iron metabolisme. Annu Rev. Nutr ., 2000, 20 , 627-662.
[36] NJAJOU O.T., VAESSEN N., JOOSSE M., BERGHUIS B., VAN DONGEN J.W., BREUNING M.H. et al. —
A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis . Nat. Genet ., 2001, 28 , 213-214.
[37] KATO J., FUJIKAWA K., KANDA M., FUKUDA N., SASAKI K., TAKAYAMA T. et al. — A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am. J. Hum. Genet ., 2001, 69 , 191-197.
[38] PIGEON C., ILYIN G., COURSELAUD B., LEROYER P., TURLIN B., BRISSOT P. et al. — A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem ., 2001, 276 , 7811-7819.
[39] NICOLAS G., BENNOUN M., DEVAUX I, BEAUMONT C., GRANDCHAMP B., KAHN A. et al. — Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (JSF2) knockout mice. Proc. Natl. Acad. Sci. U S A , 2001, 98 , 8780-8785.
[40] NICOLAS G., BENNOUN M., PORTEU A., MATIVET S., BEAUMONT C., GRANDCHAMP B. al. —
Severe iron deficiency anemia in transgenic mice expressing liver hepcidin.
Proc. Natl. Acad. Sci.
U S A , 2002, 99 , 4596-4601.
[41] ROETTO A., PAPANIKOLAOU G., POLITOU M., ALBERTI F., GIRELLI D., CHRISTAKIS J. et al. —
Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis.
Nat. Genet ., 2003, 33 , 21-22.
[42] KELLY A.L., LUNT P.W., RODRIGUES F., BERRY P.J., FLYNN D.M., Mc KIERNAN P.J. et al. —
Classification and genetic features of neonatal haemochromatosis : a study of 27 affected pedigrees and molecular analysis of genes implicated in iron metabolism. J. Med. Genet ., 2001, 38 , 599-610.
[43] GORDEUK V.R. — African iron overload.
Semin. Hematol . 2002, 39 , 263-269.
[44] BOSIO S., De GOBBI M., ROETTO A., ZECCHINA G., LEONARDO E., RIZZETTO M. et al. — Anemia and iron overload due to compound heterozygosity for novel ceruloplasmin mutations.
Blood , 2002 ; 100 , 2246-2248.
[45] BEUTLER E., GELBART T., LEE P., TREVINO R., FERNANDEZ M.A., FAIRBANKS V.F. — Molecular characterization of a case of atransferrinemia. Blood , 2000, 96 , 4071-4074.
[46] VISAPAA I., FELLMAN V., VESA J., DASVARMA A., HUTTON J.L., KUMAR V., et al. — GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am. J. Hum. Genet ., 2002, 71 , 863-876.§ [47] DE LONLAY P., VALNOT I., BARRIENTOS A., GORBATYUK M., TZAGOLOFF A., TAANMAN J.W. et al.
— A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat. Genet ., 2001, 29 , 57-60.
[48] CAMPUZANO V., MONTERMINI L., MOLTO M.D., PIANESE L., COSSÉE M., CAVALCANTI F. et al.
—Friedreich’s Ataxia : Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science . 1996, 271 , 1423-1427.
[49] MUHLENHOFF U, RICHHARDT N., RISTOW M., KISPAL G., LILL R. — The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol. Genet. 2002, 11 , 2025-2036.
[50] LESUISSE E, SANTOS R., MATZANKE B. F., KNIGHT S. A., CAMADRO J. M., DANCIS A. — Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum. Mol. Genet, 2003, 1 , 879-889.
[51] TAKETANI S., KAKIMOTO K., UETA H., MASAKi R., FURUKAWA T. — Involvement of ABC7 in the biosynthesis of heme in erythroid cells : interaction of ABC7 with ferrochelatase. Blood ., 2003, 101 , 3274-3280.
[52] HAYFLICK S.Y., WESTAWAY, S.K., LEVINSON B., ZHOU B., JOHNSON M.A., CHING K.H. et al.
—Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome.
New Engl.
J. Med ., 2003, 348 , 33-40.
[53] HÖRTNAGEL K, PROKISCH H. and MEITINGER Th. — An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum. Mol.
Genet ., 2003, 12 , 321-327.
[54] CURTIS A.R., FEY C., MORRIS C.M., BINDOFF L.A., INCE P.G., CHINNERY P. F. et al. — Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001, 28 , 350-354.
[55] BEAUMONT C., LENEUVE P., DEVAUX I., SCOAZEC J. Y., BERTHIER, M., LOISEAU M.N. et al —
Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat. Genet ., 1995, 11 , 444-446.
DISCUSSION
M.Michel BOUREL
Que dire du sigle HFE ? Quel apport à la génétique des populations face aux pourcentages décroissants du marquage génétique entre l’Europe e=du Nord-Est et la Sicile ? Le regard physiopathologique sur le métabolisme du fer ne se modifie-t-il pas, de l’entérocyte vers l’hépatocyte du fait de la mise en évidence de l’hépcidine ? Quelle place aux carences héréditaires du fer à côté des surcharges ?
Le sigle HFE, désignant le gène de l’hémochromatose, est apparu au début des années 80 dans le cadre des organisations internationales (HUGO) de cartographie du génome humain ; sa signification n’est pas évidente : peut-être faut-il retenir H pour hémochromatose et FE pour fer ? Le gradient décroissant Nord-Ouest/Sud-Est dans la fréquence de la mutation C282Y est considéré par certains comme étant la marque de l’expansion celte, par d’autres comme celle de l’expansion viking…. Quoi qu’il en soit, il paraît également nécessaire de prendre en compte non seulement la rapidité avec laquelle cette mutation s’est répandue, mais aussi la notion d’un avantage sélectif chez les sujets qui en étaient porteurs. La pathologie, les animaux invalidés et transgéniques, démontrent le rôle de la protéine HFE, de l’hepcidine (et plus récemment de l’hémojuvéline) dans la régulation de l’absorption intestinale du fer destiné à l’adapter en fonction des stocks tissulaires et des besoins de l’érythropoièse. Les mécanismes biochimiques en cause sont encore inconnus et l’une des questions soulevées est effectivement de savoir si le point de départ de cette régulation est entérocytaire ou hépatique. En pathologie humaine, et contrairement à la pathologie animale, aucune carence héréditaire en fer, responsable par exemple d’une anémie hypochrome, n’a été caractérisée. Toutes les carences sont donc considérées comme étant d’ordre nutritionnel. Il est cependant probable que différents facteurs génétiques modulent l’absorption intestinale du fer et peuvent favoriser une carence.
M. Pierre GODEAU
Chez les sujets homozygotes C282 Y n’ayant pas développé de surcharge ferrique à un âge avancé a-t-on recherché et/ou identifié un gène protecteur ?
La question de la pénétrance de l’homozygotie C282Y est extrêmement importante tant sur le plan pratique (dépistage systématique de l’hémochromatose) que conceptuelle.
L’hypothèse est effectivement faite que l’expressivité de cette homozygotie est sous la dépendance, soit d’un autre gène, soit plus vraisemblablement d’une combinaison de variants alléliques d’un certain nombre de gènes du métabolisme du fer. Différents laboratoires travaillent à l’identification de ces gènes.
M. O. ROSMORDUC
Dans le cadre de votre expérience, pourriez-vous nous indiquer la fréquence relative des mutations de ces différents gènes dans la population française ? Quel est le phénotype induit par les mutations dans le gène de la ferroportine ?
En l’état actuel des informations parues dans la littérature, les formes d’hémochromatose autres que HFE1 sont rares voire exceptionnelles. Selon toute probabilité la moins rare de ces formes est l’hémochromatose HFE4 par mutation du gène de la ferroportine (ou IREG1, ou SLC 11A3). Cette hémochromatose HFE4 présente par ailleurs trois particularités : transmission autosomique dominante, discordance entre hyperferritinémie et coefficient de saturation de la transferrine normal ou subnormal, accumulation martiale à prédominance Küpferienne.
* UMR 6061. Faculté de Médecine CS34317-2, Avenue du Pr. Léon Bernard 35043 — Rennes Cedex France et Laboratoire de Génétique Moléculaire. CHU de Rennes Tirés à part : Professeur Jean-Yves LE GALL, à l’adresse ci-dessus Article reçu le 22 avril 2003, accepté le 1er décembre 2003
Bull. Acad. Natle Méd., 2004, 188, no 2, 247-263, séance du 17 février 2004