
Résumé
La classification internationale des pneumopathies interstitielles diffuses idiopathiques publiée en 2002 comporte sept entités anatomocliniques dont le diagnostic intègre l’évaluation clinique, le scanner thoracique de haute résolution et l’aspect histopathologique pulmonaire. Elle comprend la fibrose pulmonaire idiopathique (aspect de pneumopathie interstitielle commune), la pneumopathie interstitielle non spécifique, la pneumopathie organisée cryptogénique, la bronchiolite respiratoire avec pneumopathie interstitielle diffuse, la pneumopathie interstitielle desquamative, la pneumopathie interstitielle lymphocytaire, et la pneumopathie interstitielle aiguë idiopathique (aspect de dommage alvéolaire diffus). Cette classification a permis de préciser les critères diagnostiques de chaque entité et a favorisé la recherche clinique ainsi que le développement de recommandations internationales pour la prise en charge. Une réflexion est en cours pour mieux intégrer dans la classification et la pratique le syndrome récemment identifié d’emphysème et fibrose pulmonaire combinés, les exacerbations aiguës de fibrose, ainsi que les données physiopathologiques et génétiques émergentes.
Summary
The international classification of idiopathic interstitial pneumonia published in 2002 includes seven clinical-pathologic entities distinguished by their clinical features, aspect on high-resolution computed tomography, and histopathologic findings on lung biopsy. These seven entities are idiopathic pulmonary fibrosis (with features typical of interstitial pneumonia), nonspecific interstitial pneumonia, cryptogenic organising pneumonia, respiratory bronchiolitis with interstitial lung disease, desquamative interstitial pneumonia, lymphocytic interstitial pneumonia, and acute idiopathic interstitial pneumonia (with features of diffuse alveolar damage). This classification provides clearer diagnostic criteria for each entity, has fostered clinical research and therapeutic trials, and forms the basis for international guidelines on patient care. The classification is currently being revised in order to better integrate the recently identified syndrome of combined pulmonary fibrosis and emphysema, acute exacerbations of fibrosis, and new pathophysiologic and genetic findings.
INTRODUCTION
La classification internationale des pneumopathies interstitielles diffuses (PID) idiopathiques publiée en 2002 [1, 2] comporte sept entités anatomocliniques, dont le diagnostic intègre l’évaluation clinique, le scanner thoracique de haute résolution, et l’aspect histopathologique pulmonaire. Cette classification a permis de préciser les critères diagnostiques de chaque entité et a favorisé la recherche clinique ainsi que le développement de recommandations internationales pour la prise en charge.
Les PID sont des maladies pulmonaires parenchymateuses diffuses définies sur le plan histopathologique par un processus inflammatoire et diffus, souvent fibrosant, situé de façon prédominante dans l’interstitium pulmonaire [1]. Ce dernier comprend l’interstitium alvéolaire (septums inter-alvéolaires), le tissu interstitiel souspleural et péri-bronchovasculaire, et le tissu interstitiel des parois interlobulaires. Il s’associe fréquemment à l’atteinte interstitielle des lésions des voies aériennes, parfois des alvéoles, ou de la paroi des vaisseaux. L’infiltration des structures interstitielles se traduit par des symptômes non spécifiques dominés par la dyspnée et la toux, et à l’imagerie par des opacités infiltrantes diffuses.
Sur le plan étiologique, les PID constituent un ensemble hétérogène de maladies comprenant : les PID de cause connue (médicament, antigène organique inhalé, agent minéral tel que silice ou amiante), les PID de cause inconnue survenant au cours des connectivites et maladies systémiques, les PID avec granulomatose dont la sarcoïdose, et d’autres entités bien individualisées telles que la lymphangioléiomyomatose, l’histiocytose pulmonaire langerhansienne, etc. Le cadre des PID idiopathiques est défini par l’absence d’appartenance aux groupes étiologiques ci-dessus.
HISTORIQUE
Hamman et Rich ont décrit en 1944 quatre cas de PID d’évolution aiguë, que l’on considérerait actuellement comme correspondant à une pneumopathie interstitielle aiguë idiopathique [3]. Il s’agissait d’une fibrose interstitielle diffuse pulmonaire d’installation aiguë. Une forme chronique de la maladie a été décrite dans les années 1950, et dénommée par la suite fibrose pulmonaire interstitielle chronique, ou alvéolite fibrosante cryptogénique [3]. La première classification histologique des PID a été proposée en 1968 par A.A. Liebow, et comportait alors cinq entités : pneumopathie interstitielle commune ( usual interstitial pneumonia , UIP), bronchiolite oblitérante et dommage alvéolaire diffus (BIP), pneumopathie interstitielle desquamative (DIP) [4], pneumopathie interstitielle lymphoïde (LIP), pneumopathie interstitielle à cellules géantes (GIP) [5]. La définition de chacune de ces entités a évolué par la suite, avec description de la pneumopathie organisée avec bronchiolite oblitérante (BOOP) en 1985 [6] ; de la pneumopathie interstitielle non spécifique en 1994 [7] ; et la description d’une entité radio-clinique initialement peu précise appelée fibrose pulmonaire idiopathique [8] ou alvéolite fibrosante cryptogénique, à laquelle correspondait un aspect histologique d’UIP ou de DIP. La pneumopathie interstitielle à cellules géantes a été ensuite exclue du cadre des PID idiopathiques car presque exclusivement observée lors d’une exposition aux métaux durs.
CLASSIFICATION ATS/ERS 2002
La classification actuelle des PID idiopathiques a été révisée en 2002 et fait l’objet d’un consensus international [1]. La révision de la classification était alors rendue nécessaire par l’utilisation jusque-là de plusieurs terminologies distinctes pour décrire des entités identiques comme la fibrose pulmonaire idiopathique ; l’individualisation récente de nouvelles entités anatomo-cliniques dont la pneumopathie interstitielle non spécifique [7] ; une confusion fréquente entre la terminologie histopathologique et les classifications cliniques ; et par les progrès dans la description anatomopathologique et radiologiques des PID idiopathiques, notamment grâce au recours plus fréquent à la biopsie pulmonaire depuis les progrès de la chirurgie vidéo-assistée. Dans cette classification établie par un groupe d’experts de l’American Thoracic Society (ATS) et de l’European Respiratory Society (ERS), le terme de fibrose pulmonaire idiopathique a été restreint à une entité définie sur des critères précis, et la nomenclature et les critères diagnostiques ont été standardisés [1, 9].
La classification ATS/ARS des PID idiopathiques comprend sept entités, dont six d’évolution chronique ; elle établit un lien entre chaque aspect histologique de définition précise et une entité anatomoclinique correspondante (tableau 1) [1, 10].
La fibrose pulmonaire idiopathique est la forme clinique la plus fréquente des PID idiopathiques, représentant environ 60 % des cas [1, 9]. Elle est caractérisée sur le plan histopathologique par un aspect de pneumopathie interstitielle commune (UIP), correspondant à une fibrose collagène hétérogène et mutilante de l’interstitium pulmonaire avec des foyers de prolifération fibroblastique, des lésions en rayon de miel, et peu de lésions inflammatoires [9]. La maladie survient après soixante ans, et est responsable d’une dyspnée d’effort d’installation progressive, d’une toux non productive, avec des râles crépitants bilatéraux secs des bases à l’auscultation, et fréquemment un hippocratisme digital [11]. L’évolution progressive conduit à l’insuffisance respiratoire chronique restrictive, et au décès après une durée médiane
Tableau 1. — Classification ATS/ERS 2002 des pneumopathies interstitielles diffuses idiopathiques.
Aspect histopathologique
Diagnostic clinique (approche clinique — tomodensitométrique — histopathologique)
Pneumopathie interstitielle commune Fibrose pulmonaire idiopathique Pneumopathie interstitielle non spécifique Pneumopathie interstitielle non spécifique Pneumopathie organisée Pneumopathie organisée cryptogénique Dommage alvéolaire diffus Pneumopathie interstitielle aiguë Bronchiolite respiratoire Bronchiolite respiratoire avec pneumopathie interstitielle Pneumopathie interstitielle desquamative Pneumopathie interstitielle desquamative Pneumopathie interstitielle lymphocytaire Pneumopathie interstitielle lymphocytaire de trois ans à partir du diagnostic. Le caractère idiopathique est affirmé après avoir exclu les causes connues de PID. Essentiel au diagnostic, le scanner thoracique en coupes millimétriques et haute résolution montre dans la moitié des cas au moins un aspect caractéristique qui permet alors de retenir le diagnostic, constitué d’opacités réticulaires de distribution basale et sous-pleurale prédominante, d’images pseudokystiques sous-pleurales en rayon de miel, de bronchectasies par traction, et de signes de distorsion du parenchyme pulmonaire, avec peu d’opacités en verre dépoli (Figure 1). Dans plus de la moitié des cas, le scanner thoracique permet de faire le diagnostic sans recours à la biopsie pulmonaire [12, 13]. L’exploration fonctionnelle respiratoire montre un trouble ventilatoire restrictif, une diminution de la diffusion alvéolo-capillaire, et une hypoxémie qui apparaît à l’exercice, ainsi qu’une limitation de la capacité à l’exercice [9]. Le lavage broncho-alvéolaire contribue à éliminer les diagnostics différentiels [14]. Le diagnostic repose sur l’analyse pluridisciplinaire des symptômes cliniques, du scanner thoracique, et de la biopsie pulmonaire vidéochirurgicale lorsqu’elle est indiquée [15]. Aucun traitement n’a d’efficacité dé- montrée dans cette maladie ; des essais cliniques sont en cours (bosentan, pirfénidone, macitentan, agents inhibiteurs de l’angiogénèse, etc.). Outre l’évolution habituellement lentement progressive de la maladie, il peut survenir en cours d’évolution des phases d’aggravation rapide, dites exacerbations de fibrose pulmonaire idiopathique, dont le pronostic est très mauvais et qui représentent une cause fréquente de décès [16]. Une autre complication d’identification récente est la survenue d’une hypertension pulmonaire pré-capillaire parfois grave [17].
La pneumopathie interstitielle non spécifique décrite en 1994 [7] a été peu à peu individualisée du groupe de PID idiopathiques, et en particulier distinguée de la fibrose pulmonaire idiopathique [1]. Elle est caractérisée histologiquement par l’uniformité temporelle et spatiale des lésions, la prédominance des lésions interstitielles inflammatoires par rapport à la fibrose interstitielle des cloisons alvéolaires, la préservation de l’architecture pulmonaire, et l’absence habituelle de rayon de miel et de foyer fibroblastique [7]. L’inflammation et la fibrose peuvent être présentes en proportion variable. Un aspect histologique de pneumopathie interstitielle non spécifique peut se rencontrer dans un contexte de connectivite, d’exposition environnementale à un agent organique inhalé, de prise médicamenteuse, ou dans les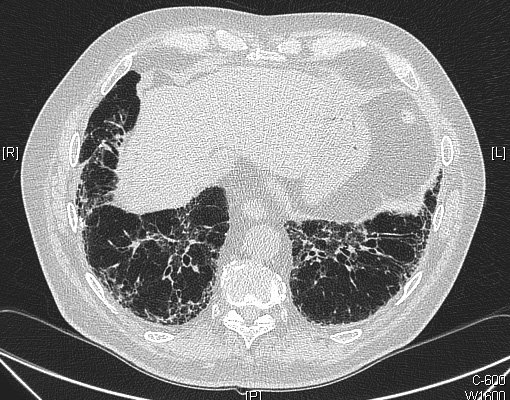
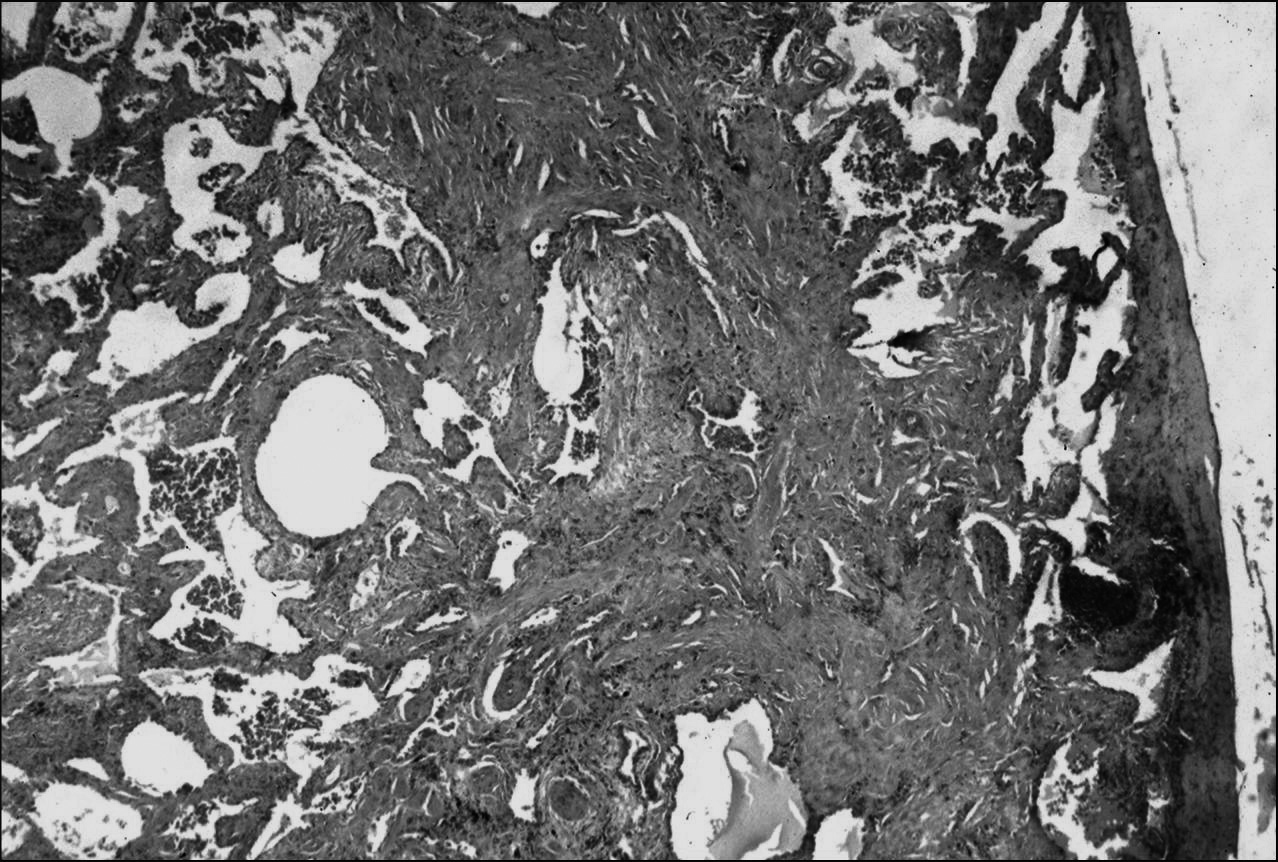
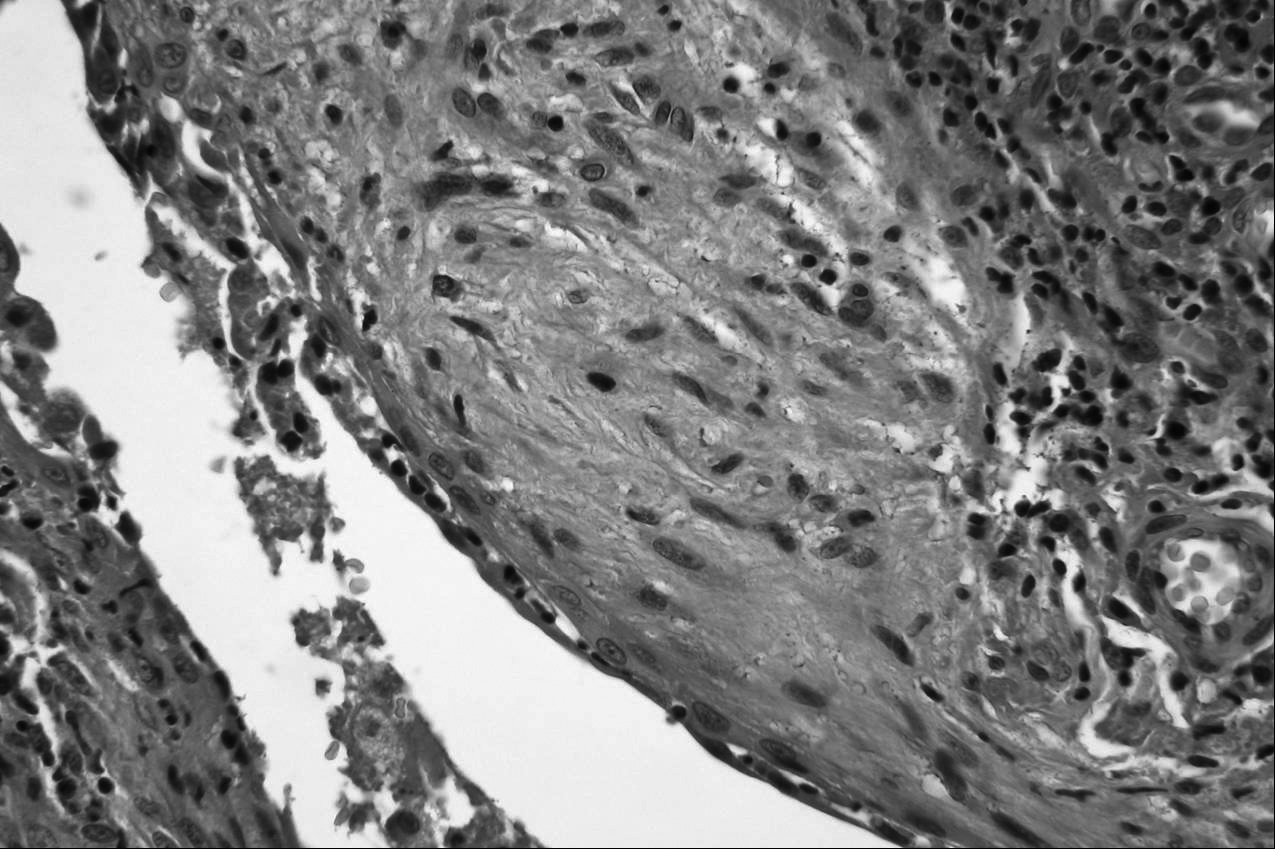
A B C Fig. 1. — Fibrose pulmonaire idiopathique. (a) Aspect tomodensitométrique: opacités réticulaires et images microkystiques sous-pleurales des bases pulmonaires en rayons de miel ; (b) aspect histopathologique à faible grossissement : hétérogénéité des lésions avec dépots de collagène et désorganisation architecturale ; (c) aspect histopathologique à fort grossissement : foyer fibroblastique.
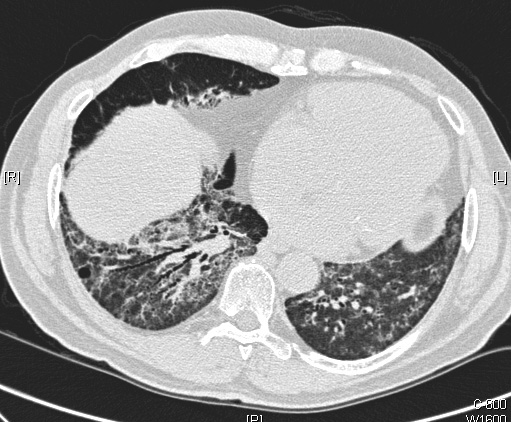
 suites d’une agression pulmonaire aiguë [7, 18] ; ces différentes causes doivent être recherchées avant de retenir le diagnostic de pneumopathie interstitielle non spécifique idiopathique. En particulier, il s’agit souvent d’une connectivite indifférenciée (fruste), dont l’installation peut être retardée [19, 20]. La présentation clinique n’est pas spécifique, mais la maladie survient en moyenne plus tôt dans la vie que la fibrose pulmonaire idiopathique (45-50 ans) ; les râles crépitants ne sont pas constants. Le lavage broncho-alvéolaire peut montrer une augmentation du pourcentage de lymphocytes et/ou des polynucléaires neutrophiles [18]. L’exploration fonctionnelle respiratoire montre des anomalies voisines de celles de la fibrose pulmonaire idiopathique. Le scanner thoracique montre le plus souvent des opacités en verre dépoli et des opacités réticulaires, souvent associées dans les mêmes territoires, ainsi que des bronchectasies par traction (Figure 2) [21]. L’aspect pseudo-kystique souspleural en rayons de miel est peu fréquent. La maladie est le plus souvent contrôlée par la corticothérapie, éventuellement associée à un traitement immunosuppresseur (azathioprine) dans les formes peu améliorées par la corticothérapie ou requérant une posologie élevée de corticoïdes sur une durée prolongée [18]. Le pronostic à long terme est nettement meilleur que celui de la fibrose pulmonaire idiopathique, avec une survie de 30 à 50 % à 10 ans [22, 23], contre 10 % pour cette dernière.
suites d’une agression pulmonaire aiguë [7, 18] ; ces différentes causes doivent être recherchées avant de retenir le diagnostic de pneumopathie interstitielle non spécifique idiopathique. En particulier, il s’agit souvent d’une connectivite indifférenciée (fruste), dont l’installation peut être retardée [19, 20]. La présentation clinique n’est pas spécifique, mais la maladie survient en moyenne plus tôt dans la vie que la fibrose pulmonaire idiopathique (45-50 ans) ; les râles crépitants ne sont pas constants. Le lavage broncho-alvéolaire peut montrer une augmentation du pourcentage de lymphocytes et/ou des polynucléaires neutrophiles [18]. L’exploration fonctionnelle respiratoire montre des anomalies voisines de celles de la fibrose pulmonaire idiopathique. Le scanner thoracique montre le plus souvent des opacités en verre dépoli et des opacités réticulaires, souvent associées dans les mêmes territoires, ainsi que des bronchectasies par traction (Figure 2) [21]. L’aspect pseudo-kystique souspleural en rayons de miel est peu fréquent. La maladie est le plus souvent contrôlée par la corticothérapie, éventuellement associée à un traitement immunosuppresseur (azathioprine) dans les formes peu améliorées par la corticothérapie ou requérant une posologie élevée de corticoïdes sur une durée prolongée [18]. Le pronostic à long terme est nettement meilleur que celui de la fibrose pulmonaire idiopathique, avec une survie de 30 à 50 % à 10 ans [22, 23], contre 10 % pour cette dernière.
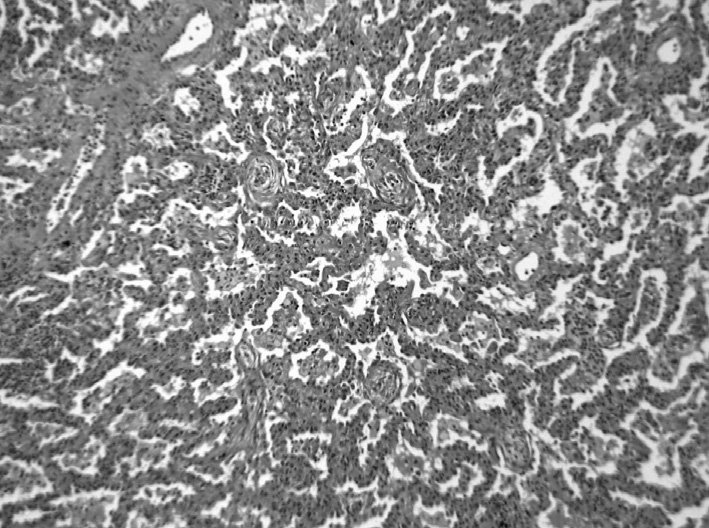
Fig. 2. — Pneumopathie interstitielle non spécifique. (a) Aspect tomodensitométrique: opacités en verre dépoli, réticulation, et bronchectasies par traction ; (b) aspect histopathologique à faible grossissement : homogénéité des lésions inflammatoires et fibreuses des cloisons alvéolaires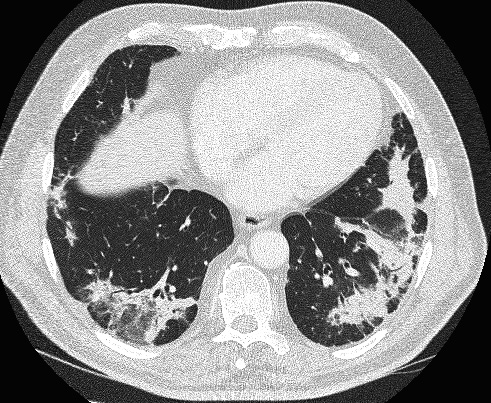
La pneumopathie organisée cryptogénique est l’entité anatomoclinique idiopathique définie par un aspect histopathologique de pneumopathie organisée, qui se traduit histologiquement par une obstruction endoluminale des espaces aériens distaux (alvéoles, canaux alvéolaires, bronchioles) par un tissu de granulation fibreux constitué de cellules inflammatoires, de fibroblastes, et de tissu conjonctif [24].
L’architecture pulmonaire est conservée. La terminologie de pneumopathie organisée a remplacé celle de bronchiolite oblitérante avec pneumopathie organisée car elle décrit mieux la prédominance de l’atteinte alvéolaire sur l’atteinte bronchiolaire et évite la confusion avec la bronchiolite oblitérante constrictive. Il ne s’agit pas à proprement parler d’une pneumopathie interstitielle, mais elle est incluse dans la classification des PID idiopathiques. L’âge moyen de survenue est de 55 ans.
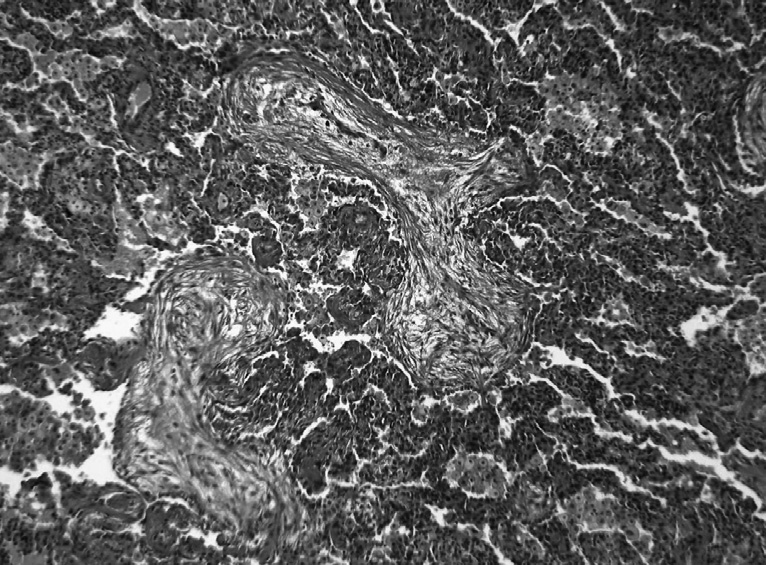
L’installation est souvent subaiguë, avec altération de l’état général, toux sèche, et dyspnée d’exercice [24]. Le retentissement fonctionnel respiratoire est le plus souvent modéré. Le lavage broncho-alvéolaire montre une augmentation des lymphocytes. L’imagerie pulmonaire montre des plages de condensation parenchymateuse avec bronchogramme aérique uni ou bilatéral (Figure 3) ; le caractère migrateur des Fig. 3. — Pneumopathie organisée cryptogénique. (a) Aspect tomodensitométrique : condensation alvéolaire bilatérale avec bronchogramme aérique ; (b) aspect histopathologique à fort grossissement ; fibrome végétant endoalvéolaire en ailes de papillons.
opacités est très évocateur du diagnostic lorsqu’il est présent [25] (il est observé également au cours de la pneumopathie chronique à éosinophiles). Dans les formes caractéristiques, un diagnostic présomptif radioclinique est parfois suffisant pour instaurer un traitement ; le diagnostic de certitude repose sur l’histologie, obtenue par biopsie pulmonaire vidéo-chirurgicale. Les biopsies transbronchiques ou la ponction biopsie pulmonaire transpariétale contribuent parfois au diagnostic [26].
De multiples causes de pneumopathie organisée doivent être recherchées avant d’admettre le caractère idiopathique [24]. Le traitement repose sur la corticothérapie par voie générale, qui est suivie d’une amélioration très rapide, avec des rechutes fréquentes.
Les autres PID, plus rares, sont détaillées ailleurs [1, 2, 10, 27]. La bronchiolite respiratoire avec pneumopathie interstitielle est une affection rare observée principalement chez les fumeurs, et caractérisée sur le plan histopathologique par la présence de macrophages (‘‘ tabagiques ’’) comportant de fines pigmentations brunes au sein des lumières des bronchioles respiratoires, des canaux alvéolaires et des espaces alvéolaires péri-bronchiolaires. Elle est peu symptomatique [28]. L’atteinte interstitielle est responsable du retentissement fonctionnel et des manifestations radiologiques, qui comportent des opacités en verre dépoli associées à un épaississement pariétal bronchique, et de multiples micronodules centrolobulaires.
La pneumopathie interstitielle desquamative est une forme rare de PID idiopathique voisine de la bronchiolite respiratoire avec PID, qui atteint préférentiellement l’adulte jeune fumeur de sexe masculin [28]. Elle est caractérisée histologiquement par l’accumulation de nombreux macrophages dans les espaces aériens distaux, et un infiltrat inflammatoire des septums inter-alvéolaires. L’imagerie montre des opacités en verre dépoli prédominant dans les bases et la région périphérique des poumons. La prise en charge repose sur l’arrêt du tabac et la corticothérapie.
L’évolution à long terme est le plus souvent favorable.
La pneumopathie interstitielle lymphocytaire [29] est une forme exceptionnelle de
PID idiopathique. Elle doit être distinguée des lymphomes B non hodgkiniens de bas grade du MALT (mu cosa associated lymphoid tissue ) par la recherche d’une clonalité lymphocytaire. La constatation d’une histologie de pneumopathie interstitielle lymphocytaire fait rechercher une connectivite, en particulier un syndrome de Gougerot-Sjögren. Le traitement repose sur la corticothérapie par voie générale.
La pneumopathie interstitielle aiguë (idiopathique) [30] est une forme rapidement progressive de PID idiopathique, dont l’histologie est décrite comme un dommage alvéolaire diffus en phase aiguë ou en voie d’organisation. Elle réalise un syndrome de détresse respiratoire aiguë de l’adulte de cause indéterminée. L’évolution vers le décès en quelques semaines survient dans la moitié des cas environ ; dans les autres cas, la maladie guérit avec peu ou sans séquelles.
CONDUITE DU DIAGNOSTIC
L’approche diagnostique des PID idiopathiques est multi-disciplinaire [15] et intègre les données cliniques, radiologiques et anatomopathologiques. Le diagnostic peut être reconsidéré tout au long de la conduite diagnostique et du suivi du patient, notamment à la faveur de la mise en évidence d’une exposition antigénique, d’une affection associée telle une connectivite, ou d’une évolution clinique discordante avec le diagnostic ou l’aspect histopathologique [2].
Le diagnostic positif de PID est porté à l’imagerie (scanner thoracique). L’évaluation clinique s’attache à identifier la présence d’une cause de la maladie, qui exclurait le caractère idiopathique de la PID. Un recueil détaillé de l’anamnèse est nécessaire en ce qui concerne les antécédents professionnels, les expositions environnementales, les prises médicamenteuses, et tout élément clinique pouvant faire suspecter une connectivite [20].
Le lavage broncho-alvéolaire est habituellement pratiqué, même si sa place dans l’algorithme diagnostique est actuellement moindre. Il permet de rechercher une cause infectieuse, une exposition pneumoconiotique, et de mettre en évidence une lymphocytose qui oriente vers un diagnostic autre que celui de fibrose pulmonaire idiopathique (pneumopathie interstitielle non spécifique ou pneumopathie organisée notamment) [14].
Le scanner thoracique de haute résolution occupe une place déterminante et croissante dans le diagnostic des PID idiopathiques. Il permet de différencier les cas où l’aspect au scanner est typique de la fibrose pulmonaire idiopathique (rayons de miel), et où la biopsie pulmonaire vidéo-chirurgicale est alors inutile, des autres présentations de PID non typiques de fibrose idiopathique (absence de rayon de miel ; opacités prédominantes en verre dépoli, distribution atypique de la maladie), où la biopsie pulmonaire est utile en l’absence de contre-indication [11]. Le scanner thoracique est moins fiable pour reconnaitre les images en rayons de miel en présence de lésions emphysémateuses associées [31]. La biopsie est toujours nécessaire pour le diagnostic précis des PID idiopathiques autres que la fibrose pulmonaire idiopathique [1]. Elle n’est toutefois pas toujours indispensable pour porter un diagnostic clinique et prendre en charge le patient. Lorsqu’elle est pratiquée, la biopsie permet d’asseoir sur des bases solides le diagnostic, l’information du patient, le pronostic, et les décisions thérapeutiques. Elle permet également d’orienter la rechercher étiologique, notamment en cas de pneumopathie interstitielle non spécifique (ou lorsqu’elle révèle un aspect histologique de pneumopathie d’hypersensibilité qui n’avait pas été suspecté avant la biopsie) [32]. La biopsie est considérée comme inutile lorsque le diagnostic radioclinique de fibrose pulmonaire idiopathique est formel, que la fibrose est avancée, et (ou) qu’il n’y a pas d’enjeu thérapeutique ; elle est déconseillée en présence de comorbidité ou d’âge physiologique avancé.
Les limites de la biopsie pulmonaire sont représentées par la difficulté d’interpré- tation des aspects histologiques atypiques, inclassables, ou les formes de chevauchement entre les différents aspects histologiques ; la modification potentielle de l’aspect histopathologique par un traitement préalable ; la possibilité d’une biopsie non représentative ; une reproductibilité imparfaite entre observateurs [33]. La distinction entre pneumopathie interstitielle commune et pneumopathie interstitielle non spécifique reste la plus difficile [34].
Au cours de l’approche diagnostique, le scanner thoracique et la biopsie pulmonaire apportent des informations pronostiques complémentaires [35]. L’approche recommandée est multi-disciplinaire, et fait participer le clinicien pneumologue, le radiologue, et l’anatomopathologiste, en particulier lorsque le diagnostic clinique est autre que celui de fibrose pulmonaire idiopathique. Il est nécessaire que cette approche soit conduite par une équipe expérimentée dans le domaine des PID.
APPORTS ET LIMITES DE LA CLASSIFICATION
Au cours des dernières années, la définition beaucoup plus précise du diagnostic de fibrose pulmonaire idiopathique, et en particulier sa distinction avec la pneumopathie interstitielle non spécifique, a permis le développement de nombreux essais thérapeutiques [36-39], faisant naître l’espoir d’améliorer le traitement de cette maladie incurable. Le progrès indéniable de cette classification a été d’homogénéiser la terminologie sur le plan international et entre les différentes spécialistes concernés (cliniciens, anatomopathologistes, radiologues), favorisant les interactions interdisciplinaires. Cette classification a également rendu possible le développement de recommandations internationales pour la prise en charge de la fibrose pulmonaire idiopathique (un document international est actuellement soumis pour publication).
La principale limite de la classification est la difficulté persistante à différencier l’aspect histopathologique de pneumopathie interstitielle commune de la pneumopathie interstitielle non spécifique [34]. Les deux aspects peuvent d’ailleurs être associés au sein de lobes différents chez un même patient. Les formes de chevauchement existent également entre la pneumopathie interstitielle desquamative et la bronchiolite respiratoire avec PID ou la pneumopathie interstitielle non spécifique.
Certaines PID restent inclassables, malgré un prélèvement de bonne taille et une analyse par des anatomopathologistes experts. Il existe également une controverse concernant le risque potentiel lié à la réalisation de la biopsie pulmonaire vidéochirurgicale ; l’analyse des données publiées permet de conclure que le risque de cet examen est faible si les contre-indications sont respectées (comorbidités importantes, âge avancé, maladie au stade d’insuffisance respiratoire chronique, fibrose pulmonaire en poussée évolutive ou exacerbation aiguë).
L’un des principes majeurs de cette classification est que le diagnostic repose sur une approche intégrée dynamique, définie par une méthode de concertation clinique, radiologique, et anatomopathologique. Cela représente un changement conceptuel majeur par rapport à l’acception préalable selon laquelle la biopsie pulmonaire chirurgicale représentait le ‘‘ gold standard ’’ du diagnostic.
PERSPECTIVES
Une révision de la classification ATS/ERS 2002 des PID idiopathiques est actuellement envisagée avec plusieurs objectifs principaux : mieux identifier d’éventuelles entités distinctes au sein de la pneumopathie interstitielle non spécifique, les patients atteints de cette affection ayant une évolution hautement variable ; intégrer à la classification les données récentes concernant les PID associées au tabagisme, et en particulier l’identification récente du syndrome d’emphysème des sommets et fibrose pulmonaire combinés [40-42] ; intégrer dans la classification l’exacerbation aiguë [16], survenant au cours de la fibrose pulmonaire idiopathique mais également d’autres PID dont la pneumopathie interstitielle non spécifique ; intégrer les données émergentes concernant les prédispositions génétiques favorisant la fibrose pulmonaire et l’existence de fibrose pulmonaire idiopathique familiale [43]. Il est ainsi possible que l’on s’oriente à l’avenir vers une classification intégrant, outre les données cliniques, l’imagerie, et l’aspect histopathologique, des informations concernant le profil moléculaire, la physiopathologie, ou les données génétiques.
L’imagerie occupant une place croissante dans l’algorithme diagnostique des PID idiopathiques et les décisions thérapeutiques dans cette maladie, sa place dans la classification des PID et l’approche pragmatique de la prise en charge doit être également reconsidérée.
REMERCIEMENTS
Madame le Professeur F. Thivolet-Béjui (Lyon) pour l’iconographie anatomopathologique ; Madame Catherine Renaud pour son aide à la préparation du manuscrit.
BIBLIOGRAPHIE [1] American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med., 2002, 165 , 277-304.
[2] Cottin V., Capron F., Grenier P., Cordier J.F. — Pneumopathies interstitielles diffuses idiopathiques. Classification de Consensus International Multidisciplinaire de l’American Thoracic Society et de l’European Respiratory Society, principales entités anatomo-cliniques, et conduite du diagnostic. Rev. Mal. Respir., 2004, 21 , 299-318.
[3] King T.E. JR, Selman M. — Introduction.
Proc. Am. Thorac. Soc., 2006, 3 , 283-284.
[4] Liebow A.A., Steer A., Billingsley J.G. — Desquamative interstitial pneumonia.
Am. J.
Med., 1965, 39 , 369-404.
[5] Liebow A. — Definition and classification of interstitial pneumonias in human pathology.
Prog. Respir. Res., 1975, 8 , 1-31.
[6] Epler G.R., Colby T.V., McLoud T.C., Carrington C.B., Geansler E.A. — Bronchiolitis obliterans organizing pneumonia. N. Engl. J. Med., 1985, 312 , 152-158.
[7] Katzenstein A.L.A., Fiorelli R.F. — Nonspecific interstitial pneumonia / fibrosis. Histological features and clinical significance. Am. J. Surg. Pathol., 1994, 18 , 136-147.
[8] Crystal R.G., Fulmer J.D., Roberts W.C., Moss M.L., Line B.R., Reynolds H.Y. — Idiopathic pulmonary fibrosis. Clinical, histologic, radiographic, physiologic, scintigraphic, cytologic, and biochemical aspects. Ann. Intern. Med., 1976, 85, 769-788.
[9] American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med., 2000, 161 , 646-664.
[10] Katzenstein A.L., Mukhopadhyay S., Myers J.L. — Erratum to ‘‘ Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases ’’ [Hum Pathol 39 (2008) 1275-1294]. Hum. Pathol., 2008, 39 , 1562-1581.
[11] Cottin V., Cordier J.F. — Fibrose pulmonaire idiopathique.
Presse Med., 2008, 37 , 1581- 1590.
[12] Hunninghake G.W., Zimmerman M.B., Schwartz D.A. et al. — Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis.
Am. J. Respir. Crit. Care Med., 2001, 164 , 193-196.
[13] Raghu G., Mageto Y.N., Lockhart D., Schmidt R.A., Wood D.E., Godwin J.D. — The accuracy of the clinical diagnosis of new-onset idiopathic pulmonary fibrosis and other interstitial lung disease: A prospective study. Chest, 1999, 116 , 1168-1174.
[14] Ohshimo S., Bonella F., Cui A. et al. — Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis.
Am. J. Respir. Crit. Care Med., 2009, 179 , 1043- 1047.
[15] Flaherty K.R., King T.E., JR., Raghu G. et al. — Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis?
Am. J. Respir. Crit. Care Med., 2004, 170 , 904-910.
[16] Collard H.R., Moore B.B., Flaherty K.R. et al. — Acute exacerbations of idiopathic pulmonary fibrosis.
Am. J. Respir. Crit. Care Med., 2007, 176 , 636-643.
[17] Lettieri C.J., Nathan S.D., Barnett S.D., Ahmad S., Shorr A.F. — Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis.
Chest, 2006, 129 , 746-752.
[18] Cottin V., Donsbeck A.V., Revel D., Loire R., Cordier J.F. — Nonspecific interstitial pneumonia. Individualization of a clinicopathologic entity in a series of 12 patients. Am. J.
Respir. Crit. Care Med., 1998, 158 , 1286-1293.
[19] Kinder B.W., Collard H.R., Koth L. et al. — Idiopathic nonspecific interstitial pneumonia:
lung manifestation of undifferentiated connective tissue disease?
Am. J. Respir. Crit. Care Med., 2007, 176 , 691-697.
[20] Cottin V. — Interstitial lung disease: are we missing formes frustes of connective tissue disease?
Eur. Respir. J., 2006, 28 , 893-896.
[21] Elliot T.L., Lynch D.A., Newell J.D. JR. et al. — High-resolution computed tomography features of nonspecific interstitial pneumonia and usual interstitial pneumonia.
J. Comput.
Assist. Tomogr., 2005, 29 , 339-345.
[22] Travis W.D., Matsui K., Moss J., Ferrans V.J. — Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns. Survival comparison with usual interstitial pneumonia and desquamative intersitial pneumonia. Am. J. Surg. Pathol., 2000, 24 , 19-33.
[23] Daniil Z.D., Gilchrist F.C., Nicholson A.G. et al. — A histological pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am. J. Respir. Crit. Care Med., 1999, 160 , 899-905.
[24] Cordier J.F. — Cryptogenic organising pneumonia.
Eur. Respir. J., 2006, 28 , 422-446.
[25] Cordier J.F., Loire R., Brune J. — Idiopathic bronchiolitis obliterans organizing pneumonia.
Definition of characteristic clinical profiles in a series of 16 patients. Chest, 1989, 96 , 999-1004.
[26] Cottin V. — Progrès pour le diagnostic de pneumopathie organisée.
Rev. Mal. Respir., 2008, 25 , 523-525.
[27] Leslie K.O. — My approach to interstitial lung disease using clinical, radiological and histopathological patterns. J. Clin. Pathol., 2009, 62 , 387-401.
[28] Ryu J.H., Myers J.L., Capizzi S.A., Douglas W.W., Vassallo R., Decker P.A. — Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease.
Chest, 2005, 127 , 178-184.
[29] Swigris J.J., Berry G.J., Raffin T.A., Kuschner W.G. — Lymphoid interstitial pneumonia:
a narrative review. Chest, 2002, 122 , 2150-2164.
[30] Vourlekis J.S. — Acute interstitial pneumonia.
Clin. Chest Med., 2004, 25 , 739-747, vii.
[31] Akira M., Inoue Y., Kitaichi M., Yamamoto S., Arai T., Toyokawa K. — Usual interstitial pneumonia and nonspecific interstitial pneumonia with and without concurrent emphysema:
thin-section CT findings. Radiology, 2009, 251 , 271-279.
[32] Vourlekis J.S., Schwarz M.I., Cool C.D., Tuder R.M., King T.E., JR., Brown K.K. — Nonspecific interstitial pneumonitis as the sole histologic expression of hypersensitivity pneumonitis. Am. J. Med., 2002, 112 , 490-493.
[33] Flaherty K.R., Travis W.D., Colby T.V. et al. — Histopathologic variability in usual and nonspecific interstitial pneumonias.
Am. J. Respir. Crit. Care Med., 2001, 164 , 1722-1727.
[34] Du Bois R., King T.E. JR. — Challenges in pulmonary fibrosis × 5 : the NSIP/UIP debate.
Thorax, 2007, 62 , 1008-1012.
[35] Flaherty K.R., Thwaite E.L., Kazerooni E.A. et al. — Radiological versus histological diagnosis in UIP and NSIP: survival implications.
Thorax, 2003, 58 , 143-148.
[36] Raghu G., Brown K.K., Costabel U. et al. — Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial.
Am. J. Respir. Crit. Care Med., 2008, 178 , 948-955.
[37] King T.E., JR., Behr J., Brown K.K. et al. — BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis.
Am. J. Respir. Crit. Care Med., 2008, 177 , 75-81.
[38] King T.E., JR., Albera C., Bradford W.Z. et al. — Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebocontrolled trial. Lancet, 2009, 374 , 222-228.
[39] Taniguchi H., Ebina M., Kondoh Y. et al. — Pirfenidone in idiopathic pulmonary fibrosis.
Eur. Respir. J., 2009 (sous presse).
[40] Cottin V., Nunes H., Brillet P.Y. et al. — and the Groupe d’Etude et de Recherche sur les
Maladies ‘‘ Orphelines ’’ Pulmonaires (GERM ’’O ’’P). Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur. Respir. J., 2005, 26 , 586-593.
[41] Cottin V., Brillet P.Y., Nunes H., Cordier J.F. — Syndrome d’emphysème des sommets et fibrose pulmonaire des bases combinés. Presse Med., 2007, 36 , 936-944.
[42] Cottin V., Le Pavec J., Prévot G. et al. — Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome.
Eur. Respir. J, 2010, 35 , 105-111.
[43] Steele M.P., Speer M.C., Loyd J.E. et al. — Clinical and pathologic features of familial interstitial pneumonia.
Am. J. Respir. Crit. Care Med., 2005, 172 , 1146-1152.
DISCUSSION
M. François-Bernard MICHEL
Votre travail d’expert peut-il bénéficier, en réseau, au pneumologue qui, constatant sur un scanner des lésions de fibrose, éprouve des difficultés à aller plus loin dans la caractérisation ?
Les pneumopathies interstitielles diffuses sont des maladies pulmonaires rares (prévalence inférieure à 1/2 000 habitants). À ce titre, leur prise en charge relève du réseau de Centres de Compétence pour les maladies pulmonaires rares, coordonné par le Centre National de Référence (coordonnateur Jean-François Cordier). Les Centres de Compé- tence et le Centre de Référence ont établi des collaborations avec des anatomopathologistes, des radiologues, et des chirurgiens thoraciques, ayant une expertise de ce groupe de pathologies pour une approche multidisciplinaire, et travaillent en lien étroit avec les pneumologues hospitaliers et libéraux.
M. Jacques BATTIN
Ayant observé et publié une communication sur le syndrome de Hamman et Rich mortel dans la première année de vie, de tels cas aussi exceptionnellement précoces sont-ils connus, sont-ils des sous diagnostiqués et sont-ils expliqués par des mutations dominantes ?
Les pneumopathies interstitielles idiopathiques existent chez l’enfant, même si elles sont plus rares et moins étudiées. Il existe des formes familiales de pneumopathie interstitielle diffuse idiopathique, pouvant prendre suivant les sujets d’une même famille un aspect histopathologique de pneumopathie interstitielle commune, de pneumopathie interstitielle non spécifique, ou même de pneumopathie interstitielle desquamative. Dans ces formes familiales ont été rapportées des mutations des gènes codant pour les protéines du surfactant, et plus récemment des mutations des gènes du complexe télomérase, certaines d’entre elles étant dominantes.
M. Christian NEZELOF
Puis-je vous interroger sur le caractère familial de certaines fibroses pulmonaires idiopathiques et rappeler qu’il existe chez l’enfant, une pneumonie interstitielle à plasmocytes, à l’origine de laquelle une cause est parfois retrouvée ?
Il existe en effet des formes familiales de pneumopathie interstitielle diffuse idiopathique, ainsi que des fibroses pulmonaires idiopathiques familiales. La pneumopathie interstitielle à plasmocytes est maintenant considérée comme synonyme de la pneumopathie interstitielle lymphocytaire ; les contextes étiologiques les plus fréquents sont ceux de l’infection par le virus de l’immunodéficience humaine et du syndrome de GougerotSjögren, mais des formes idiopathiques sont décrites. Le diagnostic différentiel avec les lymphomes pulmonaires est difficile.
M. Jacques ROCHEMAURE
Devant les progrès de l’imagerie, le lavage alvéolaire est-il toujours indispensable devant une fibrose interstitielle évidente, sachant que le traitement cortisonique sera souvent tenté ?
Le lavage alvéolaire est utile dans le cadre du diagnostic étiologique, pour éliminer les causes infectieuses de pneumopathies interstitielles, faire une recherche minéralogique, rechercher une lymphocytose (qui oriente vers une sarcoïdose, une pneumopathie d’hypersensibilité, plus rarement une pneumopathie médicamenteuse ; et dans le cadre des pneumopathies interstitielles diffuses idiopathiques vers une pneumopathie interstitielle non spécifique, une pneumopathie organisée, ou une pneumopathie interstitielle lymphocytaire). Toutefois, le lavage alvéolaire ne permet pas le diagnostic de certitude dans le cadre des pneumopathies interstitielles diffuses idiopathiques, et sa place dans la conduite du diagnostic est actuellement moindre.
M. Jacques-Louis BINET
Dans les formes lymphocytaires, s’agit-il de lymphocytes B puisque vous avez souvent observé la présence d’une immunoglobuline monoclonale ?
Les pneumopathies interstitielles lymphocytaires comportent en effet une prédominance de lymphocytes B. Il est utile de rechercher une clonalité pour le diagnostic différentiel avec le lymphome pulmonaire.
Mme Dominique LECOMTE
Nous observons très souvent des pneumopathies interstitielles fibrosantes diffuses ou en foyers chez des sujets décédés dont la vie était très précaire. Avez-vous la notion d’un lien entre fibrose et vie précaire ?
Il n’y a pas à ma connaissance de données épidémiologiques associant fibrose pulmonaire diffuse et conditions socio-économiques défavorables. Mais un aspect localisé de fibrose pulmonaire peut représenter la séquelle d’une agression pulmonaire non spécifique ancienne, fréquente chez ces patients.
M. Charles-Joël MENKÈS
La pneumopathie interstitielle lymphocytaire peut-elle être distinguée des localisations pulmonaires de la polyarthrite rhumatoïde ?
La pneumopathie interstitielle de la polyarthrite rhumatoïde prend le plus souvent l’aspect histopathologique de la pneumopathie interstitielle commune, ou de la pneumopathie interstitielle non spécifique, les autres aspects histopathologiques étant plus rares ;
des lésions de bronchiolite sont très souvent associées.
* Hospices civils de Lyon, Hôpital Louis Pradel, Service de pneumologie et Centre de référence des maladies pulmonaires rares. Université de Lyon, Université Lyon 1, UMR 754, 28 avenue Doyen Lépine — 69677 Bron ; e-mail : vincent.cottin@chu-lyon.fr Tirés à part : Professeur Vincent Cottin, même adresse Article reçu le 15 décembre 2009, accepté le 18 janvier 2010
Bull. Acad. Natle Méd., 2010, 194, no 2, 327-342, séance du 2 février 2010