
Résumé
Nous présentons les constatations histologiques des biopsies nerveuses de six enfants souffrant d’une neuropathie héréditaire sensitivo-motrice d’évolution sévère, en rapport avec des mutations du gène de la mitofusine 2. Les études électrophysiologiques sont en faveur d’une atteinte axonale qui est confirmée sur la biopsie nerveuse par une raréfaction des fibres myélinisées. Dans tous les cas, il existe des lésions des mitochondries, bien mises en évidence sur les sections longitudinales. Ces structures sont anormalement sphériques, plus petites que la normale et regroupées. Nous avons confronté ces constatations à l’étude de plusieurs nerfs contrôles. Il est possible que ces anomalies morphologiques mitochondriales soient liées à des anomalies de la fusion — fission mitochondriale. Dans différentes formes de maladie de Charcot-Marie-Tooth, induites par des mutations des gènes PMP22, MPZ, GJB1, GDAP1, MTMR2, SH3TC2, PRX, FGD4, LMNA, il existe des signes histologiques qui permettent d’orienter vers des mutations de l’un de ces gènes. Ceci parait également vrai pour le gène MFN2.
Summary
We present neuropathological findings based on sural nerve biopsy in six children with mutations of the mitofusin 2 gene (MFN2). All six children had severe axonal neuropathies (mild or severe hereditary motor and sensory neuropathy, HMSN), with onset in early childhood. All had a marked decrease in the density of mainly large myelinated fibers. Although neurophysiological findings were suggestive of axonal degeneration, some onion bulbs were present in each case. Unequivocal mitochondrial changes were apparent only on longitudinal sections. Many axonal mitochondria appeared smaller than normal and round or spherical instead of tubular. These mitochondria were abnormally aggregated, accumulating primarily at the axon periphery. This peripheral distribution was clearest in residual large myelinated fibers. The inner and outer mitochondrial membranes were irregular, and the cristae were quite often disrupted. These changes were observed in both myelinated and unmyelinated fibers. Mitofusin 2 is a large mitochondrial transmembrane GTPase, with two coiled coil domains and two transmembrane spans. It is targeted to the outer mitochondrial membrane, where it interacts with mitofusin 1 to regulate the mitochondrial network architecture by stimulating mitochondrial fusion. The mitochondrial changes we observed could thus result from abnormal mitochondrial fusion and fission. Neuropathologic abnormalities can be sufficiently characteristic to suggest the genetic basis of some hereditary neuropathies such as those associated with mutations in MPZ, GJB1, GDAP1, MTMR2, SH3TC2, PRX, FGD4 and LMNA. This may also be true of MFN2-related neuropathies.
INTRODUCTION
Une forme clinique de neuropathie sensitivo-motrice sévère débutant tôt dans l’enfance a été décrit en 1981 par Ouvrier et coll . : « early onset axonal neuropathy »
EOAN [1]. Le phénotype clinique est sévère et évolue rapidement, si bien que la plupart des patients présentent une atteinte motrice distale très sévère, invalidante, au moment de l’adolescence. Les constatations électrophysiologiques et neuropathologiques sont plutôt en faveur d’une axonopathie [2].
Les ADN de huit cas cliniquement typiques, sporadiques, dont sept avaient été rapportés par Ouvrier et coll . en 1981, ont été étudiés à la recherche d’une éventuelle anomalie génique ; pour six d’entre eux, nous avons pu mettre en évidence des mutations du gène codant pour la mitofusine 2 ( MFN2 ) (MIM : 608507). Il a été prouvé récemment que des mutations dominantes de ce gène étaient effectivement susceptibles d’expliquer des formes axonales de CMT : CMT2A, et plus particuliè- rement des formes de début précoce [3, 4].
Nous présentons ici les anomalies microscopiques observées sur les biopsies nerveuses de ces six patients ; de tels examens, en particulier ultrastructuraux, n’ont été réalisés que très rarement chez des patients présentant une mutation de MFN2 .
MÉTHODES
Tous les patients ont été examinés de façon détaillée, cliniquement et par des études électroneuromyographiques. Les biopsies des nerfs suraux de ces six patients EOAN, ont été réalisées après consentement éclairé des parents des patients ou des patients eux-mêmes, dont la neuropathie a débuté avant l’âge de cinq ans. Les âges à la première biopsie étaient les suivants : cas 1, six ans ; cas 2, deux ans ; cas 3, huit ans ; cas 4, six ans ; cas 5, trois ans ; cas 6, trois ans. Une deuxième biopsie fut réalisée chez trois malades : cas 1, treize ans ; cas 5, quatorze ans ; cas 6, vingt-deux ans.
L’examen a été systématiquement réalisé en microscopie optique et électronique.
Pour chaque prélèvement, un fragment a été inclus en paraffine et examiné en microscopie optique ; un autre a été fixé dans le glutaraldéhyde, inclus dans l’épon et préparé pour la microscopie optique (coupes semi-fines), puis pour la microscopie électronique. Afin d’étudier au mieux les mitochondries axonales, sur chaque biopsie furent réalisées de nombreuses sections longitudinales qui ont été examinées de façon approfondie en microscopie électronique et comparées à des sections longitudinales de nerfs contrôles normaux ou anormaux sélectionnés au hasard dans notre laboratoire parmi des biopsies nerveuses réalisées depuis trente ans :
— adultes : trois cas de neuropathie héréditaire sensitivo-motrice liée à une mutation du gène de la lamine A/C (CMT2B1 — forme axonale autosomique récessive), trois cas de polyradiculonévrite inflammatoire démyélinisante chronique, une polyneuropathie associée à une insuffisance rénale, une polyneuropathie axonale de cause inconnue, quatre nerfs normaux, — enfants : trois nerfs normaux chez des sujets âgés respectivement de huit mois, cinq ans et neuf ans.
Nous avons par ailleurs réalisé une étude quantitative des fibres myélinisées ainsi qu’une étude systématique de la longueur des mitochondries sur les sections longitudinales en microscopie électronique. Les données des patients et des contrôles ont été comparées en utilisant le test de Mann-Whitney.
L’analyse moléculaire du gène MFN2 a été réalisée par séquençage direct. Les 19 exons (incluant les deux premiers exons non codant) ont été amplifiés par PCR en utilisant des amorces oligonucléotidiques introniques (séquences des amorces disponibles sur demande). Les deux brins d’ADN ont été séquencés à l’aide du Big Dye Terminator Cycle Sequencing Kit v.1.1 (Applied Biosystems) sur un séquenceur automatique (ABI3130xl, Applied Biosystems). La recherche de mutation a été réalisée à l’aide du logiciel Sequence Navigator version 1.0.1 (Applied Biosystems).
RÉSULTATS
Les neuropathies périphériques des six patients rapportés ici étaient en rapport pour l’un avec une mutation homozygote de MFN2 , pour deux avec des mutations
Tableau 1. — Mutations de
MFN2 identifiées chez nos patients
CAS 1 [c.647T>C] + [c.647T>C] ;
[p.Phe216Ser]+ [p.Phe216Ser] 2 c.310C>T ; p.Arg104Trp 3 c.310C>T ; p.Arg104Trp 4 [c.640G>A] + [c.1168T>C] ;
[p.Asp214Asn] + [Cys390Arg] 5 c.292A>G ; p.Lys98Glu 6 [c.491C>T] + [c.1085C>T] ;
[p.Ala164Val] + [p. Thr362Met] hétérozygotes composites (affectant les deux allèles du gène) et pour les trois derniers avec des mutations hétérozygotes (tableau 1).
Chez tous les patients a été observée une très significative diminution de la densité des fibres myélinisées concernant de façon prédominante celles de grand diamètre.
Entre les deux biopsies réalisées chez les cas 1, 5 et 6, a pu être observée une diminution significative de la densité des fibres myélinisées avec simplement persistance des fibres de petit diamètre. Un nombre significatif des gaines de myéline de ces fibres restantes étaient plus fines que la normale. Quelques bouquets de régéné- rescence ont été observés ; un nombre significatif de proliférations schwanniennes en bulbes d’oignon, entourant habituellement des gaines de myéline trop fines par rapport au diamètre axonal (cas 3 et 4), a été mis en évidence par l’examen en microscopie électronique.
Sur toutes les biopsies ont été constatées des lésions des mitochondries à l’examen de sections longitudinales. Beaucoup de ces mitochondries axonales semblaient plus petites que la normale, rondes et sphériques au lieu d’être tubulaires. Elles étaient anormalement agrégées et s’accumulaient de façon focale, surtout à la périphérie des axones lorsqu’il s’agissait d’axones de grand diamètre, alors que ces agrégats pouvaient occuper tout le diamètre des fibres amyéliniques qui ont un petit diamè- tre. Une quantification de la taille des mitochondries axonales (mesure de la longueur du plus grand axe de toutes les mitochondries visibles) a mis en évidence une diminution très significative (p<0.001 dans tous les cas) de la taille des mitochondries dans les nerfs de tous nos patients par comparaison à des nerfs normaux provenant de sujets de même âge : longueur moyenne des mitochondries comprise entre 0,40 et 1,06 μm chez les patients, entre 2,20 et 3,15 μm chez les témoins.
Les membranes interne et externe des mitochondries étaient irrégulières et souvent les crêtes étaient désorganisées. Aucune inclusion paracristalline n’a été observée.
Ces aspects anormaux n’ont pas paru plus fréquents au niveau des nœuds de
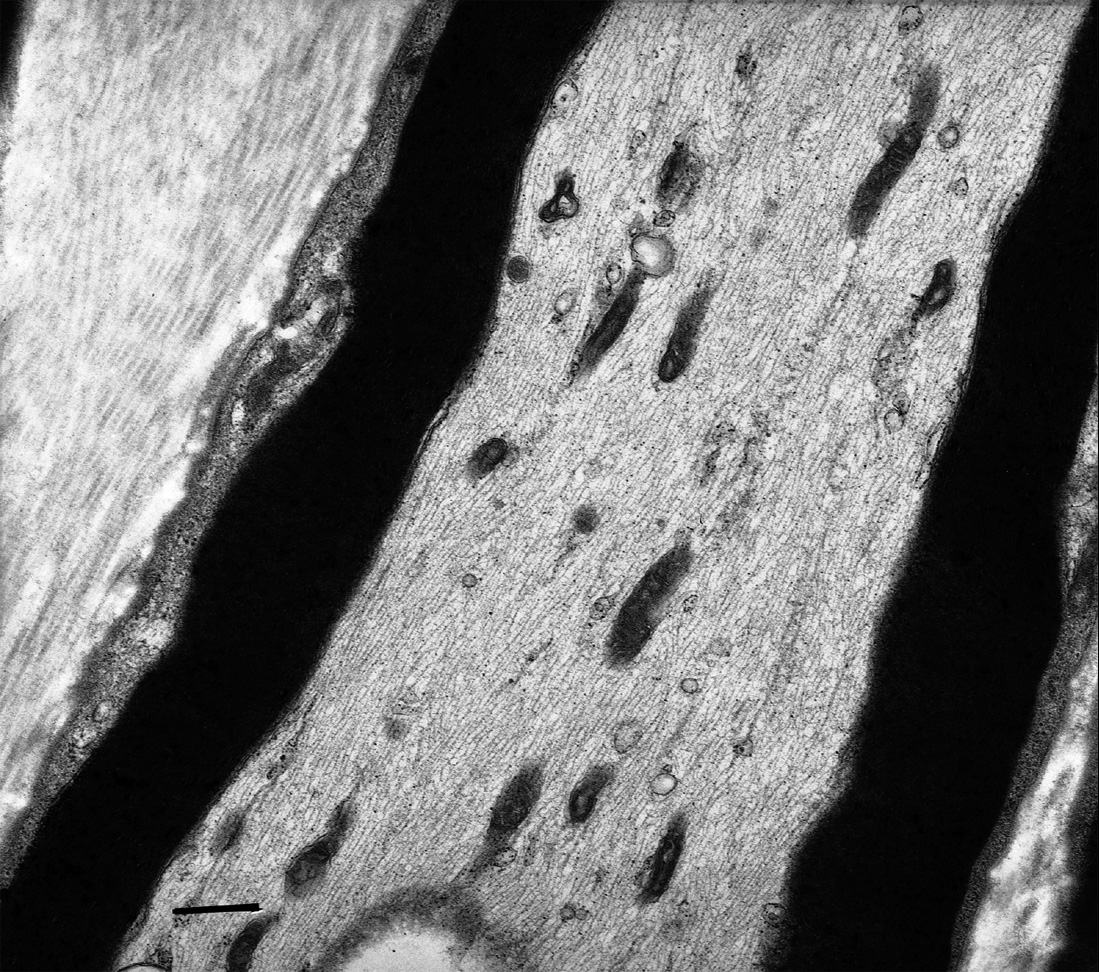
Fig. 1. — Micrographie électronique. Nerf normal. Section longitudinale. Des mitochondries tubulaires sont alignées de façon aléatoire dans le sens d’un axone. Barre = 500nm.
Fig. 2. — Micrographie électronique. Cas 4. Section longitudinale. Des mitochondries anormales sont regroupées, de forme plus ou moins sphérique et présentent des anomalies de leurs parois et de leurs crêtes. Barre = 200 nm.
Ranvier. Dans tous les cas néanmoins, était aussi observé un nombre significatif d’axones dans lesquels les mitochondries étaient d’aspect normal. Chez les patients ayant subi deux biopsies, la perte axonale (mesurée par la densité des fibres myélinisées) était plus importante lors de la deuxième biopsie, mais les anomalies mitochondriales ne paraissaient pas plus importantes. Nous n’avons pas constaté de lésion particulière des mitochondries des cellules de Schwann, des cellules périneurales, des cellules endothéliales, et les autres composants axonaux étaient normaux.
DISCUSSION
Trois de nos patients (cas 2, 3 et 5) portaient une mutation hétérozygote du gène MFN2 (tableau 1). Des mutations hétérozygotes ont été trouvées dans un nombre significatif de familles atteintes de forme axonale de CMT (CMT2A) (10 à 30 % des cas). De plus, des mutations hétérozygotes de novo ont été décrites chez plusieurs patients présentant une forme sporadique de neuropathie de début très précoce avec parfois une atrophie optique [5-8]. Chez le patient 2, l’obtention d’échantillons d’ADN provenant des deux parents asymptomatiques a permis de confirmer qu’il s’agissait bien d’une mutation de novo. Par ailleurs, les cas 1, 4 et 6 présentaient une mutation sur les deux allèles du gène MFN2 . Chez trois patients présentant une telle neuropathie sporadique de début précoce avec mutation des deux allèles de
MFN2 (incluant les cas 4 et 6 de cette série), nous avons pu constater que chaque mutation était héritée d’un parent asymptomatique n’ayant aucune atteinte électrophysiologique du système nerveux périphérique ce qui indique que la transmission dans ces cas était récessive. Il apparaît donc que le phénotype de neuropathie axonale de début très précoce observé chez nos patients définit une catégorie particulière de CMT2A de transmission soit dominante, soit récessive.
La mutation dominante présente chez les cas 2 et 3 (p.Arg104Trp) ainsi qu’une des deux mutations récessives du patient 6 (p.Thr362Met) ont déjà été rapportées précédemment [2, 9]. Les autres mutations n’ont jamais été rapportées auparavant mais touchent des acides aminés très conservés au cours de l’évolution et n’ont pas été observées chez cent sujets contrôles. On peut noter que les deux mutations observées chez le cas 6 (p.Ala164Val et p.Thr362Met) ne sont pas responsables de changements importants en termes de charge, d’hydrophobicité ou d’encombrement stérique. Il est probable que seule l’association de ces deux mutations d’effet modéré peut entraîner un phénotype (pas ou peu de symptômes à l’état hétérozygote).
Les anomalies histologiques étaient identiques dans tous les cas de notre série. Il s’agissait d’une perte sévère des grandes fibres myélinisées, de quelques bouquets de régénérescence de fibres myélinisées et chez deux patients, d’une prolifération significative des cellules de Schwann en bulbes d’oignon. Récemment, Lacour et coll. ont rapporté un cas de neuropathie liée à une mutation de MFN2 qui présentait des caractères très démyélinisants sur le plan électrophysiologique et histologique [10]. En ce qui concerne la dégénérescence axonale et la démyélinisation, il convient de souligner que la perte axonale est un élément constant de toute neuropathie d’évolution chronique progressive, cette notion expliquant les anomalies axonales constatées à l’examen électroneuromyographique de tous les malades. Néanmoins, cette atteinte axonale ne préjuge pas du mécanisme initial qui pourrait être démyé- linisant ou axonal.
En ce qui concerne les anomalies mitochondriales, Chung et coll. ont présenté les résultats des biopsies de sept patients, mais n’ont pas décrit d’anomalie des mitochondries [4]. Verhoeven et coll. sont les seuls à avoir rapporté des anomalies mitochondriales sur des sections transversales étudiées en microscopie électronique de nerfs suraux de deux enfants qui avaient une mutation de MFN2 [3]. Ces auteurs observent des modifications dégénératives des mitochondries axonales, décrites comme petites, denses aux électrons chez un malade et dilatées chez l’autre. Ils décrivent également des anomalies des crêtes mitochondriales et dans un cas, une accumulation focale de mitochondries en voie de dégénérescence.
En fait, notre étude souligne la nécessité de l’étude des sections longitudinales en microscopie électronique qui permet d’apprécier au mieux la morphologie des mitochondries. L’agrégation anormale de ces mitochondries rondes est très caractéristique, tandis que chez les contrôles les mitochondries sont dispersées de façon aléatoire dans les axones. Nous avons également observé des irrégularités des membranes mitochondriales interne et externe et de sévères anomalies des crêtes. Il faut néanmoins souligner que les mitochondries sont des structures très fragiles et sensibles à la fixation et que du fait de ce risque d’artefacts des anomalies des crêtes doivent donc être interprétées avec prudence. De plus, la confirmation de la spécificité de ces anomalies morphologiques ultrastructurales viendra de l’étude d’autres biopsies nerveuses contrôles dans des conditions techniques identiques.
La mitofusine 2 (Mfn2) est une GTPase transmembranaire mitochondriale qui est localisée sur la membrane mitochondriale externe où elle interagit avec la mitofusine 1 (Mfn1) pour réguler l’architecture du réseau mitochondrial en stimulant la fusion des mitochondries. Les deux protéines Mfn1 et Mfn2 forment des complexes homo-oligomériques et hétéro-oligomériques qui sont essentiels pour la fusion mitochondriale. En fait, un équilibre entre les phénomènes de fusion et de fission détermine la morphologie mitochondriale normale, à savoir l’aspect tubulaire caractéristique sur les sections longitudinales des axones. Les anomalies mitochondriales que nous avons décrites sont donc suggestives d’un défaut de fusion mitochondriale secondaire à une dysfonction de Mfn2. Des précédentes études dans des modèles cellulaires de CMT2A ont déjà suggéré une telle dysfonction : Baloh et coll.
ont en effet exprimé les mutations de MFN2 rencontrées au cours du CMT2A dans des cultures de neurones de ganglions rachidiens postérieurs et ont observé des regroupements anormaux de petites mitochondries fragmentées dans les corps neuronaux déficients en Mfn2 [11]. Chen et coll. ont observé en microscopie électronique que les mitochondries, dans de telles cultures, étaient sphériques ou ovalaires et de taille variable [12]. Par ailleurs, des données récentes suggèrent que les processus de fusion-fission sont non seulement importants pour maintenir la morphologie normale des mitochondries mais aussi leur fonction dans les cellules. Baloh et coll. ont également signalé que dans un modèle cellulaire les mutations de MFN2 entraînaient un ralentissement de leur transport axonal, dont l’intensité pourrait être responsable de la sévérité du phénotype [11]. Nous n’avons cependant pas observé de diminution évidente de la densité mitochondriale dans les nerfs suraux biopsiés chez nos patients, et n’avons donc pas de preuve de l’intervention de ce mécanisme dans les neuropathies périphériques humaines dues à des mutations de MFN2 .
D’un point de vue génétique, la constatation de telles anomalies chez des patients présentant des mutations soit récessives, soit dominantes, suggère que la plupart des mutations faux-sens de MFN2 sont responsables d’une perte de fonction conduisant à une fusion mitochondriale anormale. Il est probable que chez des sujets dont la transmission de la mutation est dominante, la présence d’une Mfn2 normale n’est pas capable de compenser le déficit dû à la mutation responsable du phénotype clinique que nous avons décrit. Une étude récente vient à l’appui de cette hypothèse, en montrant que dans des cellules déficientes soit en Mfn1 soit en Mfn2 et dans les quelles on exprime une protéine Mfn2 mutée, la forme normale de Mfn2 ne peut pas complémenter la Mfn2 mutée, tandis que la forme normale de Mfn1 en est capable (dans le premier cas, les mitochondries restent fragmentées tandis que dans le deuxième cas la forme tubulaire des mitochondries est restaurée) [13]. La complé- mentation par une Mfn1 normale fait intervenir des complexes hétérooligomériques entre Mfn2 mutée et Mfn1 normale. En revanche, les complexes homo-oligomériques entre Mfn2 normale et mutée sont non fonctionnels pour la fusion mitochondriale et ne peuvent restaurer la tubulation mitochondriale. Il a été suggéré que les nerfs périphériques pourraient ne contenir que très peu ou pas de Mfn1, ne permettant pas la compensation de la Mfn2 mutée. Cette dysfonction de la dynamique mitochondriale couplée à l’extrême longueur des axones impliqués pourrait être responsable de la dégénérescence axonale observée dans ces neuropathies [14]. Cependant l’expression de Mfn1 dans le nerf périphérique n’a jamais été testée ni comparée à celle de Mfn2, et nécessitera des investigations complémentaires sur des nerfs normaux et lésés pour vérifier expérimentalement cette hypothèse.
CONCLUSION
Nous concluons donc de nos observations et de la littérature que dans ce contexte de neuropathie axonale sévère chronique de l’enfant, la présence de mutations du gène MFN2 induit des anomalies mitochondriales détectables par un examen ultrastructural.
Plus de vingt-cinq gènes responsables de différentes formes de maladie de CharcotMarie-Tooth ont été identifiés à ce jour. Chez certains patients, des lésions ultrastructurales caractéristiques peuvent être utiles pour suggérer des mutations de tel ou tel gène :
PMP22 (nombreux bulbes d’oignon en rapport avec une prolifération concentrique de cellules de Schwann autour d’une fibre démyélinisée ou en voie de remyélinisation), GJB1 (nombreux bouquets de régénérescence), MTMR2,
MTMR13 et FGD4 (grand nombre de proliférations aberrantes des gaines de myéline),
SH3TC2 (anomalies évocatrices des fibres amyéliniques), PRX (modifications discrètes de la myéline des nœuds de Ranvier) et
LMNA (raréfaction sévère des fibres myélinisées sans signe de régénérescence). Nous estimons donc que la constatation, à l’examen en microscopie électronique de sections longitudinales de biopsies nerveuses, d’anomalies mitochondriales peut orienter vers une mutation du gène MFN2 . Il pourrait être intéressant de réaliser une étude similaire de biopsies nerveuses de patients atteints d’une maladie de CMT induite par une mutation du gène GDAP1 (CMT4A), puisque la protéine codée par ce gène est également impliquée dans les phénomènes de fusion-fission mitochondriale.
Il convient de rappeler ici que l’indication d’une biopsie nerveuse est discutée au cas pour cas et n’est proposée qu’aux patients pour lesquels les données cliniques, familiales et les explorations moléculaires n’ont pas permis de déterminer la mutation causale dans un gène en particulier.
BIBLIOGRAPHIE [1] Ouvrier R.A., McLeod J.G., Morgan G.J., Wise G.A., Conchin T.E. — Hereditary motor and sensory neuropathy of neuronal type with onset in early childhood. J. Neurol. Sci. , 1981, 51 , 181-197.
[2] Nicholson G.A., Magdelaine C., Zhu D. et al. — Severe early-onset axonal neuropathy with homozygous and compound heterozygous
MFN2 mutations. Neurology , 2008, 70 , 1678-1681.
[3] Verhoeven K., Claeys K.G., Züchner S. et al. — MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2.
Brain , 2006, 129 , 2093-2102.
[4] Chung K.W., Kim S.B., Park K.D. et al. — Early onset severe and late-onset mild Charcot-
Marie-Tooth disease with mitofusin 2 (
MFN2 ) mutations. Brain , 2006, 129 , 2103-2113.
[5] Züchner S., Mersiyanova I.V., Muglia M. et al. — Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A.
Nat Genet, 2004, 36 ,449-451.
[6] Züchner S., De Jonghe P., Jordanova A. et al. — Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2.
Ann Neurol, 2006, 59 , 276-281.
[7] Lawson V.H., Graham B.V., Flanigan K.M. — Clinical and electrophysiologic features of CMT 2A with mutations in the mitofusin 2 ( MFN2 ) gene. Neurology , 2005, 65 , 197-204.
[8] Neusch C., Senderek J. Eggermann T. Elolff E., Bahr M, Schneider-Gold C. — Mitofusin 2 gene mutation (R94Q) causing severe early-onset axonal polyneuropathy (CMT2A). Eur. J.
Neurol. , 2007, 14 , 575-577.
[9] Brockmann K., Dreha-Kulaczewski S., Dechent P. et al. — Cerebral involvement in axonal
Charcot-Marie-Tooth neuropathy caused by mitofusin 2 mutations.
J. Neurol. , 2008, 255 , 1049-1058.
[10] Lacour A., Srojkovic T., Latour P., Zephir H., Hurtevent J.F., Wang A. — Un cas de neuropathie démyélinisante inhomogène lié au gène MFN2 . Rev. Neurol. , 2007, 163 (suppl. no 4) , M11 p. 2S115.
[11] Baloh R.H., Schmidt R.E., Pestronk A., Milbrandt J. — Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J.
Neurosci ., 2007, 27 , 422-430.
[12] Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. — Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell. Biol. , 2003, 160 , 189-200.
[13] Detmer S.A., Chan D.C. — Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A mutations. J. Cell. Biol., 2007, 176 , 405-414.
[14] Detmer S.A., Chan D.C. — Functions and dysfunctions of mitochondrial dynamics.
Nat. Rev.
Mol. Cell. Biol. , 2007, 8 , 870-879.
DISCUSSION
M. Jean-Marie Léger
Existe-t-il d’autres exemples remarquables de corrélations avec les anomalies morphologiques observées sur les biopsies neuro-musculaires, dans d’autres variétés de maladie de Charcot-Marie Tooth ?
Il convient tout d’abord de rappeler que la biopsie nerveuse est un examen invasif, dont l’indication doit être posée avec discernement, notamment quand les examens de première ligne n’ont pas permis d’aboutir au diagnostic étiologique de la neuropathie. Dans le cas des neuropathies génétiquement déterminées de type Charcot-Marie-Tooth, les premières analyses de biologie moléculaire sont presque toujours réalisées avant que soit posée l’indication d’une biopsie nerveuse. Cela étant dit, il existe effectivement d’autres exemples d’anomalies de la biopsie nerveuse qui vont faire suspecter la présence de mutations dans un gène spécifique ; un exemples caractéristique est celui du gène MPZ , codant pour la protéine P0, dont les mutations vont causer une décompaction de la gaine de myéline visible en microscopie électronique.
M. Raymond Ardaillou
Quelles sont les conséquences de l’invalidation de la mitofusine 2 chez la souris ? Est-il possible d’invalider spécifiquement ce gène dans le système nerveux ?
L’invalidation des gènes
Mfn1 ou Mfn2 à l’état homozygote chez la souris est responsable d’une létalité embryonnaire. Il a cependant été possible de cultiver des fibroblastes provenant de ces embryons et donc porteurs d’une invalidation homozygote de Mfn1 ou
Mfn2 . Ces fibroblastes embryonnaires présentent des anomalies morphologiques tout à fait marquées des mitochondries, qui apparaissent courtes et arrondies au lieu d’avoir leur forme tubulaire habituelle. Il est intéressant de noter que ces cellules peuvent être complémentées par transfert de la forme sauvage de la protéine non déficitaire (c’est-à- dire tranfert d’un transgène codant pour Mfn1 chez les souris invalidées pour Mfn2, ou d’un transgène codant pour Mfn2 chez les souris invalidées pour Mfn1). Dans ce cas le phénotype mitochondrial est en partie corrigé, montrant qu’un effet de dosage génique contribue au phénotype mitochondrial de ces fibroblastes embryonnaires. Concernant la deuxième question, une invalidation du gène Mfn2 a déjà été réalisée dans le cervelet de souris par l’équipe de David Chan (California Institute of Technology, USA), permettant de montrer un lien entre déficit de fusion mitochondriale et dégénérescence des cellules de Purkinje. En revanche, ce type d’invalidation génique n’a jamais été réalisé dans le système nerveux périphérique. L’équipe de David Chan a cependant réalisé une expérience un peu différente, en créant des souris transgéniques exprimant une protéine Mfn2 porteuse d’une mutation ponctuelle responsable de maladie de Charcot-MarieTooth. A l’état homozygote (double dose de transgène), les souris transgéniques présentent des symptômes évocateurs d’une neuropathie périphérique. Les nerfs de ces souris n’ont malheureusement pas été étudiés en microscopie électronique pour l’instant.
* Membre correspondant de l’Académie nationale de médecine ** Service de Neurologie et Centre de Référence des Neuropathies périphériques rares, CHU Dupuytren — Limoges — France *** Laboratoire de Biochimie et de Génétique Moléculaire, CHU Dupuytren — Limoges — France **** Institut de Recherche Neuromusculaire, Children’s Hospital, Westmead — Australie Tirés à part : Professeur Jean-Michel Vallat, service et laboratoire de neurologie, 2 avenue Martin Luther King, CHU Dupuytren 87042 Limoges, France. Article reçu le 3 novembre 2008, accepté le 12 janvier 2009
Bull. Acad. Natle Méd., 2009, 193, no 1, 151-161, séance du 13 janvier 2008