
Résumé
Le développement d’une fibrose hépatique repose sur l’accumulation progressive de constituants de la matrice extra-cellulaire pouvant conduire à la destruction de la structure du parenchyme hépatique. Cette accumulation provient de la formation de fibroblastes activés en réponse à des agressions virales, auto-immunes, toxiques ou mécaniques, qui vont produire de manière incontrôlée les composants de la matrice, tels que des collagènes de types I, III et V et des molécules de fibronectine. Les cellules à l’origine de ces fibroblastes activés constituent un sujet d’intenses recherches. Alors qu’il est admis que les cellules étoilées du foie participent de façon très significative à la génération de ces cellules fibrogéniques, il est apparu plus récemment que d’autres cellules du parenchyme hépatique pouvaient également jouer un rôle significatif. En particulier, des travaux récents ont proposé l’implication des hépatocytes au travers de la réactivation aberrante d’un phénomène de transdifférenciation cellulaire embryonnaire appelé transition épithéliomésenchymateuse. Les observations ayant conduit à cette hypothèse encore controversée seront présentées et les limites des approches utilisées seront discutées.
Summary
Liver fibrosis is due to excessive deposition of extracellular matrix by fibroblasts that are actived in response to chronic liver injury, including viral, autoimmune, toxic and mechanical stresses. These cells produce many of the constituents of the matrix, including type I, type II and type V collagen and fibronectin. However, the precise origin of these fibroblasts is controversial. Quiescent hepatic stellate cells are generally believed to be the main source of fibrogenic cells. but accumulating evidence suggests that they are not the sole culprits. It was recently proposed that hepatocytes may undergo an embryonic transdifferentiation process, known as epithelial-mesenchymal transition, that allows them to acquire a fibroblastic phenotype during liver fibrosis. Experimental observations underlying this controversial hypothesis are presented and the limits of current methodological approaches are discussed.
La fibrose hépatique est caractérisée par une accumulation excessive et un remodelage de la matrice extracellulaire (MEC) conduisant à une altération structurale et fonctionnelle du foie. De manière similaire aux fibroses affectant d’autres tissus, tels que les poumons, le rein, le cœur ou le tissu cutané, la fibrose hépatique provient d’un déséquilibre entre la synthèse des constituants normaux de la matrice, leur dépôt et leur dégradation. La MEC est constituée d’un assemblage complexe de macromolécules incluant les collagènes, en particulier de types I et III, les glycoprotéines, l’élastine, les protéoglycanes et les glycosaminoglycanes. Il s’agit d’un tissu en remodelage constant au sein duquel les cellules peuvent migrer et interagir. Dans le foie normal, la production (fibrogenèse) et la dégradation (fibrolyse) de la matrice sont en équilibre. Sous l’effet répété des agressions virales, auto-immunes, biliaires, toxiques ou mécaniques, les hépatocytes, les cellules endothéliales, les cellules de Kupffer ou les lymphocytes vont sécréter des cytokines qui vont entraîner la transformation des cellules quiescentes du foie en myofibroblastes caractérisés par la présence de filaments d’alpha-actine de type musculaire lisse. Ces cellules vont produire des protéines de la matrice et des collagènes en quantité anormale, aboutissant à une accumulation excessive de la matrice au dépend du parenchyme hépatique ainsi qu’à une distribution anormale des composants matriciels dans des territoires du parenchyme hépatique qui en sont normalement dépourvus.
Les cellules à l’origine des myofibroblastes activés semblent varier selon l’étiologie et du type de fibrose. Ainsi, il est généralement admis que dans le cas des fibroses d’origine métabolique avec distribution péri-sinusoïdale les myofibroblastes dérivent des cellules étoilées du foie (anciennement appelées cellules de Ito), alors que dans le cas des fibroses vasculaires et des des fibroses cicatricielles ils dériveraient de fibroblastes présents en région péri-portale ou centro-lobulaire [1-8].
Cette distinction est cependant restrictive. Les myofibroblastes activés constituent en effet une population cellulaire hétérogène au sein d’une même lésion [9], certains pouvant dériver des cellules étoilées du foie et d’autres de cellules mésenchymateuses. De plus, les hépatocytes, les cellules épithéliales biliaires et les cellules endothéliales bordant les sinusoïdes hépatiques peuvent participer à la fibrogenèse en produisant certains constituants de la MEC. L’hypothèse selon laquelle plusieurs types cellulaires participent à l’accumulation excessive de MEC au cours de la fibrogenèse a été confortée récemment par la mise en évidence d’un processus de transition épithélio-mésenchymateuse (TEM) dans des modèles animaux de fibrose hépatique [10].
La TEM est un processus de transdifférenciation permettant de générer des cellules de phénotype mésenchymateux à partir d’un épithélium structuré. Elle se traduit par une perte des jonctions intercellulaires, une dépolarisation des cellules, l’induction de l’expression de métalloprotéases, une destruction de la membrane basale et l’invasion des tissus adjacents [11]. Il s’agit d’un processus morphogénique fondamental impliqué, notamment, dans la mise en place du mésoderme au cours de la gastrulation, de la migration des cellules de la crête neurale, de la formation des valves cardiaques et de la fusion du palais chez les vertébrés supérieurs [11]. Dans le foie fœtal, les hépatocytes sont observés dans divers stades de différenciation intermédiaire avec co-expression de marqueurs mésenchymateux et de marqueurs d’hépatocytes, suggérant que pendant le développement embryonnaire, les hépatocytes peuvent contribuer à l’émergence de cellules mésenchymateuses au travers d’une TEM [12, 13]. La réactivation de ce processus embryonnaire a été impliquée chez l’adulte dans deux conditions pathologiques : les fibroses et les processus de cancérogenèse [11, 14]. Cette réactivation est la conséquence de l’induction de l’expression de facteurs de transcription embryonnaires contrôlant ce phénomène de transdifférenciation (tels que les facteurs Snail, Twist et Zeb), en réponse à des blessures ou à des conditions de stress cellulaires [15].
C’est en 2007 que l’équipe de Rhagu Kalluri a présenté les premières observations expérimentales suggérant l’implication d’une transdifférenciation d’hépatocytes en fibroblastes activés au cours de la fibrogenèse hépatique sur la base de l’analyse d’un modèle murin [10]. Au bout de six semaines de traitement par injection intrapéritonéale de térachlorure de carbone (CCl ), les souris développent une fibrose hépati4 que caractérisée par l’accumulation de deux populations de fibroblastes. La première population est caractérisée par l’expression de la protéine αSMA, marqueur classique des fibroblastes activés dérivant des cellules étoilées du foie, alors que la deuxième population de fibroblastes est caractérisée par l’expression de la protéine FSP1, protéine liant le calcium de la famille des protéines S100. De façon intéressante, ce dernier sous-type de fibroblastes peut également être mis en évidence après traitement in vitro d’hépatocytes murins différenciés par le TGFβ, une cytokine capable d’induire une TEM (Figure 1). Ces observations suggéraient donc qu’en réponse au CCl , deux populations de fibroblastes participent à la fibrogenèse : une 4 population dérivée des cellules étoilées du foie, et une population générée à partir des hépatocytes par un processus de TEM et caractérisée par l’expression de FSP1.
Avec l’objectif de valider cette hypothèse, les auteurs ont développé un modèle de souris transgénique permettant de suivre le devenir des hépatocytes au cours de la fibrogenèse. Pour ce faire, ils ont d’abord généré une lignée de souris présentant l’expression d’une ADN recombinase (la recombinase Cre) sous le contrôle du promoteur du gène de l’albumine, gène exprimé de façon spécifique par les hépatocytes (souris Albumine — Cre ; Figure 2A). Chez ces souris la recombinase Cre n’est donc exprimée que dans les hépatocytes. Les auteurs de cette étude ont ensuite créé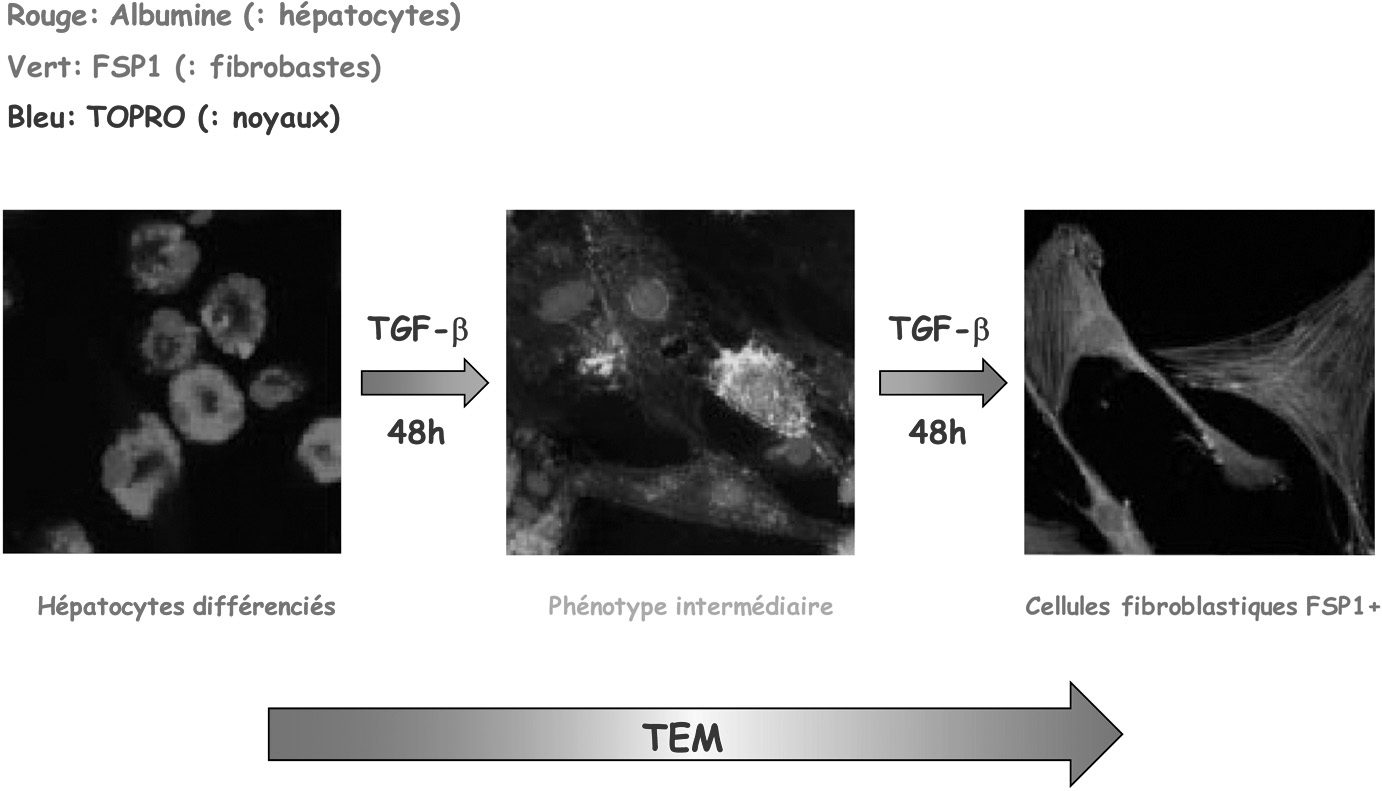
Fig. 1. —
In vitro , le TGF-β (3 ng/ml) induit la formation de fibroblastes exprimant la protéine FSP1 à partir d’hépatocytes différenciés [10]. TEM : transition épithélio-mésenchymateuse.
une deuxième lignée de souris transgéniques présentant un gène marqueur (le gène LacZ , codant pour la β-galactosidase) sous le contrôle d’un promoteur ubiquitaire (souris Rosa26 — STOP —
LacZ ; Figure 2A). Cependant, l’expression du marqueur est bloquée par la présence d’un codon STOP placé en amont de la séquence LacZ . Ce codon est entouré par deux sites LoxP, sites de reconnaissance de la recombinase. Ils ont ensuite croisé ces souris pour obtenir des animaux doubletransgéniques (souris Rosa26 — STOP — LacZ ; Albumine — Cre). Dans les hépatocytes de ces souris la recombinase Cre est exprimée et peut ainsi exciser le codon STOP. Le gène marqueur LacZ est alors exprimé dans les hépatocytes conduisant à l’accumulation de la β-galactosidase. Cette expression peut être mise en évidence sur coupe histologique en utilisant un substrat chromogénique de la β-galactosidase : le X-gal, à l’origine d’une couleur bleue (Figure 2B). Ce même marqueur se trouvant alors sous le contrôle d’un promoteur ubiquitaire, son expression est maintenue quel que soit le devenir des cellules. Après traitement de ces souris double-transgéniques par le CCl , deux types de fibroblastes, positifs pour 4 αSMA et positifs pour FSP1, sont observés dans les lésions hépatiques, confortant l’hypothèse de l’implication de plusieurs types cellulaires. L’expression du gène LacZ est détectée dans une proportion significative des fibroblastes positifs pour
FSP1 (Figure 2C), indiquant une origine hépatocytaire. Confirmant l’implication du processus de TEM dans l’initiation et la progression de la fibrose, l’administration d’un inhibiteur de la voie du TGF-β, une protéine recombinante BMP7 (Bone Morphogenic-protein 7) permet d’inhiber la TEM et de freiner l’évolution de la lésion. Ces observations présentaient des implications théoriques et pratiques
Fig. 2. — Suivi in vivo du devenir des hépatocytes différenciés au cours de la fibrogenèse selon l’équipe de Raghu Kalluri [10].
A. Description des modèles animaux utilisés. Génération des souris double-transgéniques (dT) :
Rosa26 — STOP — LacZ ; Albumine — Cre.
B. Mise en évidence des hépatocytes et des cellules dérivées des hépatocytes dans le foie des souris double-transgéniques.
C. Mise en évidence dans les régions fibreuses du foie des souris double-transgéniques traitées par le CCl de cellules fibroblastiques dérivant d’hépatocytes.
4 importantes pour l’étude des fibroses, la survenue d’une TEM pouvant être à l’origine d’une mobilisation rapide de cellules fibrogéniques dans la zone lésée. Elles ouvraient également de nouvelles perspectives thérapeutiques, par inhibition des voies de signalisation contrôlant ce processus de transdifférenciation.
Alors que les observations de l’équipe de R. Kalluri semblaient indiquer l’origine hépatocytaire d’une sous-population de fibroblastes présents dans la zone de fibrose, elles restaient insuffisantes pour démontrer leur intervention directe dans la fibrogenèse, aucune donnée ne montrant que ces fibroblastes participaient à la sécrétion de composants de la matrice extra-cellulaire. Afin de tester l’implication pathogénique de ces fibroblastes, l’équipe de David Brenner [16] a généré en 2010 des souris triple-transgéniques en croisant les souris précédemment développées par l’équipe de R. Kalluri (souris Rosa26 — STOP — LacZ ; Albumine — Cre) avec des souris transgéniques présentant un gène codant pour un marqueur fluorescent (la protéine GFP) sous le contrôle du promoteur du gène codant pour un collagène de type I (collagène 1α) (Figure 3A). Chez ces souris triple-transgéniques (Rosa26 — STOP — LacZ ; Albumine — Cre ; Col I — GFP) seules les cellules produisant du collagène 1α sont donc fluorescentes. Dans le foie de ces souris, la présence de fibroblastes dérivés d’hépatocytes exprimant le gène du collagène 1α se traduit donc par l’expression du gène de la β-galactosidase ( LacZ ), signant l’origine hépatocytaire, et de la protéine fluorescente, signant sa capacité à exprimer le collagène 1α.
Après traitement des souris par le CCl , les chercheurs de cette équipe ont donc 4 analysé les différentes sous-populations de fibroblastes présents dans les régions fibrotiques. En contradiction avec les résultats obtenus par l’équipe de R. Kalluri, aucune cellule exprimant la β-galactosidase, c’est-à-dire d’origine hépatocytaire, ne présentait de marqueurs fibroblastiques, que ce soit la protéine αSMA ou la protéine FSP1 (Figure 3B). L’absence de cellules exprimant de façon conjointe la β-galactosidase et la protéine GFP reflétait également leur incapacité à exprimer et donc à secréter du collagène 1α. Suite à la publication de ces données contradictoires, l’implication de la TEM a été également remise en cause dans d’autres types de fibroses, en particulier dans la fibrose biliaire et la fibrose rénale [17-19]. Alors que de multiples observations in vitro et in vivo démontrent l’implication de ce phénomène de transdifférenciation cellulaire au cours de la tumorigenèse et de la progression tumorale [20, 21], son intervention dans les fibroses tissulaires reste donc à démontrer.
Au-delà de la nécessité d’améliorer la connaissance de la physiopathologie des fibroses tissulaires, ces résultats contradictoires nous amènent à nous interroger sur les limites de certaines approches expérimentales. Il est en effet troublant de constater que l’utilisation de modèles identiques peut conduire à des conclusions opposées. A ce jour, l’explication la plus probable de cette contradiction repose sur le manque de spécificité des approches analytiques et des marqueurs utilisés par l’équipe de R. Kalluri [22]. Si le développement de la biologie moléculaire au cours de ces 20 dernières années a conduit à des avancées technologiques déterminantes, en particulier dans le domaine de la transgenèse avec le développement de modèles
Fig. 3. — Caractérisation in vivo du devenir des hépatocytes différenciés au cours de la fibrogenèse selon l’équipe de David Brenner [16].
A.Description des modèles animaux utilisés. Génération des souris triple-transgéniques (tT) :
Rosa26 — STOP — LacZ ; Albumine — Cre ; Col I — GFP.
B. Analyse des hépatocytes et des cellules dérivées des hépatocytes dans le foie des souris tripletransgéniques.
C. Mise en évidence dans les régions fibreuses du foie des souris double-transgéniques traitées par le CCl de cellules fibroblastiques dérivant d’hépatocytes.
4 animaux de plus en plus sophistiqués, ces résultats contradictoires nous rappellent avec force l’importance de disposer de marqueurs parfaitement caractérisés et de méthodologies standardisées afin d’assurer la qualité des analyses des données obtenues et d’éviter des conclusions parfois hâtives.
BIBLIOGRAPHIE [1] Friedman S.L. — Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem , 2000, 275 , 2247-2250.
[2] Bataller R., North K.E., Brenner D.A. — Genetic polymorphisms and the progression of liver fibrosis: a critical appraisal. Hepatology , 2003, 37 , 493-503.
[3] Li D., Friedman S.L. — Liver fibrogenesis and the role of hepatic stellate cells: new insights and prospects for therapy. J. Gastroenterol. Hepatol ., 1999, 14 , 618-633.
[4] Gabele E., Brenner D.A., Rippe R.A. — Liver fibrosis: signals leading to the amplification of the fibrogenic hepatic stellate cell. Front Biosci ., 2003, 8 , D69-77.
[5] Brenner D.A., Waterboer T., Choi S.K. et al . — New aspects of hepatic fibrosis. J. Hepatol ., 2000, 32 , 32-38.
[6] Knittel T., Kobold D., Saile B. et al . — Rat liver myofibroblasts and hepatic stellate cells:
different cell populations of the fibroblasts lineage with fibrogenic potential.
Gastroenterology , 1999, 117 , 1205-1221.
[7] Mehal W.Z. — Activation-induced cell death of hepatic stellate cells by the innate immune system. Gastroenterology , 2006, 130 , 600-603.
[8] Pavlova A., Stuart R.O., Pohl M. et al . — Evolution of gene expression patterns in a model of branching morphogenesis.
Am. J. Physiol ., 1999, 277 , F650-F663.
[9] Magness S.T., Bataller R., Yang L. et al . — A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations.
Hepatology , 2004, 40 , 1151- 1159.
[10] Zeisberg M., Yang C., Martino M. et al . — Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition.
J. Biol. Chem. , 2007, 282 , 23337-23347.
[11] Thiery J.P., Acloque H., Huang Y.J.H. et al . — Epithelial-mesenchymal transitions in development and disease.
Cell , 2009, 139 , 871-890.
[12] Chagraoui J., Lepage-Noll A., Anjo A. et al . — Fetal liver stroma consists of cells in epithelial-to-mesenchymal transition.
Blood , 2003, 101 , 2973-2982.
[13] Pagan R., Martin I., Llobera M. et al . — Epithelial-mesenchymal transition of cultured rat neonatal hepatocytes is differentially regulated in response to epidermal growth factor and dimethyl sulfoxide. Hepatology , 1997, 25 , 598-606.
[14] Iwano M., Plieth D., Danoff T.M. et al . — Evidence that fibroblasts derive from epithelium during tissue fibrosis.
J. Clin. Invest ., 2002, 110 , 341-350.
[15] Thiery J.P., Sleeman J.P. — Complex networks orchestrate epithelial-mesenchymal transitions.
Nat. Rev. Mol. Cell Biol ., 2006, 7 , 131-142.
[16] Taura K., Miura K., Iwaisako K. et al . — Hepatocytes do not undergo epithelialmesenchymal transition in liver fibrosis in mice.
Hepatology , 2010, 51 , 1027-1036.
[17] Chu A.S., Diaz R., Hui J.J. et al . — Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis.
Hepatology , 2011, 53 , 1685-1695.
[18] Scholten D., Osterreicher C.H., Scholten A. et al . — Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice.
Gastroenterology , 2010 , 139 , 987-998.
[19] Humphreys B.D., Lin S.L., Kobayashi A. et al. — Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis.
Am. J. Pathol ., 2010, 176, 85-97.
[20] Ansieau S., Morel A.P., Hinkal G. et al . — Twisting an embryonic factor into an oncoprotein.
Oncogene , 2010, 29, 3173-3184.
[21] Puisieux A. — Role of epithelial-mesenchymal transition in tumour progression.
Bull. Acad.
Natl Med. , 2009, 193, 2017-2032.
[22] Wells R.G. — The epithelial-to-mesenchymal transition in liver fibrosis: here today, gone tomorrow? Hepatology , 2010, 51 , 737-740.
* Membre correspondant de l’Académie nationale de médecine, Cancérologie — Lyon Inserm UMR 10512, CNRS UMR 5286 et Centre Léon Bérard ; e-mail : alain.puisieux@lyon.unicancer.fr Tirés à part : Professeur Alain Puisieux, même adresse