
Résumé
C’est au début du siècle dernier que les principales régions cérébrales responsables de la veille et du sommeil sont identifiées au travers d’études associant sections/lésions cérébrales et analyse des états de vigilance. Dans les décennies suivantes, l’identification neurochimique des populations neuronales sous-tendant la régulation des états de vigilance, ainsi que la caractérisation de leurs liens anatomiques et fonctionnels ont permis de modéliser les mécanismes de genèse de l’éveil et du sommeil. De fait, ce modèle est basé sur l’existence d’interactions inhibitrices réciproques entre deux types de neurones dont le profil d’activité varie en fonction des états de vigilance : les neurones de l’éveil principalement monoaminergiques et cholinergiques, et les neurones du sommeil dont les neurones GABAergiques de l’aire préoptique de l’hypothalamus. Outre les neuromédiateurs de ces systèmes, en particulier la sérotonine, l’acétylcholine et le GABA, de nouvelles molécules neuroactives ont été découvertes récemment, permettant de revisiter le modèle des mécanismes de régulation des états de vigilance. C’est tout particulièrement le cas de l’hypocrétine (ou orexine), un neuropeptide impliqué dans une pathologie du sommeil, la narcolepsie. La caractérisation des rôles respectifs de ces différents neuromédiateurs a permis l’identification de nouvelles cibles thérapeutiques pour le traitement des troubles du sommeil tels que les récepteurs à hypocrétine dont le blocage pharmacologique a des propriétés hypnotiques.
Summary
By combining brain section/lesion studies and sleep analysis, neurophysiologists have identified the brain areas responsible for regulating sleep and wakefulness during the first half of the 20th century. Identification of the phenotypic nature of the neurons underlying the regulation of vigilance, as well as their anatomical and functional connections led to a theoretical model based on mutual inhibitory interactions between sleep-on neurons (namely GABAergic neurons of the hypothalamic preoptic region) and wake-on neurons (mainly monoaminergic and cholinergic neurons). In addition to the corresponding neurotransmitters (serotonin, acetylcholine and GABA), other neuroactive molecules that play key roles in sleep and wakefulness regulation have recently been discovered, leading to an updated model. Hypocretin, also known as orexin, is a key neuropeptide involved in the sleep disorder narcolepsy. Extensive characterization of the respective roles of these neurotransmitters has led to the identification of novel therapeutic targets for the treatment of sleep disorders. For example, blockade of hypocretin receptors has hypnotic effects.
INTRODUCTION
Chez les mammifères, les états de vigilance se décomposent en trois stades, éveil, sommeil lent et sommeil paradoxal, qui alternent selon une organisation propre à chaque espèce et se définissent par des activités corticales et musculaires parfaitement caractérisées (Figure 1). Les troubles du sommeil affectent un nombre important de personnes : 30 % de la population générale se plaindrait de son sommeil. Les données des études épidémiologiques montrent que la prévalence des pathologies les plus fréquentes se situe entre 6 et 15 % pour l’insomnie, entre 2 et 4 % pour le syndrome d’apnées du sommeil et est de l’ordre de 10 % pour le syndrome des jambes sans repos [1]. Dans ce contexte, la compréhension des mécanismes qui contrôlent les états de vigilance s’avère un enjeu d’importance pour appréhender les processus physiopathologiques à l’origine des troubles du sommeil et proposer de nouvelles stratégies thérapeutiques.
Historique : découverte des régions cérébrales responsables de l’éveil
Au début du siècle dernier, deux régions cérébrales sont identifiées comme centres de l’éveil: l’hypothalamus et le tronc cérébral.
En 1918, les pays européens sont touchés par une épidémie d’encéphalite virale caractérisée par un état quasi-permanent de somnolence. Constantin von Economo, un neurologue viennois, fut le premier à identifier la cause de cette léthargie : la lésion de l’hypothalamus postérieur [2]. Par ailleurs, certains patients souffraient au contraire d’insomnie permanente et l’examen post mortem montrait alors une lésion de l’hypothalamus antérieur. De ces observations, Von Economo émit l’hypothèse que des centres spécifiques d’éveil et du sommeil étaient localisés dans l’hypothalamus. Cette découverte a ensuite été confirmée par des expériences de lésions chez l’animal [3].
Fig. 1. — Les trois principaux états de vigilance chez la souris.
Chez les mammifères, les états de vigilance se définissent par trois variables physiologiques: le tonus musculaire évalué par l’électromyogramme (EMG), les mouvements des yeux suivis par l’électrooculogramme (EOG) et l’activité corticale objectivée sur l’électroencéphalogramme (EEG). Sur la base de ces caractéristiques polygraphiques, on distingue trois états de vigilance : l’éveil, le sommeil lent et le sommeil paradoxal.
L’ éveil est associé à un tonus musculaire élevé et de nombreux mouvements oculaires. L’EEG d’éveil se caractérise par une activité électrique corticale dite désynchronisée ou activée, avec des signaux à haute fréquence et de faible amplitude (bas voltage). Le sommeil lent se définit par une diminution du tonus musculaire et des ondes lentes synchronisées sur l’EEG. Enfin, le sommeil paradoxal se caractérise par une atonie musculaire illustrée sur l’EMG, des mouvements oculaires rapides visibles à l’EOG et une activité corticale proche de celle observée pendant l’éveil sur l’EEG.
Dans les années 1930, l’importance du tronc cérébral dans l’activation corticale a été suggérée par les données du neurophysiologiste Frédéric Bremer [4]. Ce dernier, grâce à l’enregistrement de l’activité électrophysiologique du cortex cérébral chez le chat, a montré qu’une transsection isolant le cerveau antérieur du tronc cérébral (préparation dite « cerveau isolé ») induisait un sommeil lent permanent, alors qu’une transsection plus postérieure, en arrière du tronc cérébral (préparation dite « encéphale isolé »), préservait une alternance veille/sommeil (Figure 2). Bremer en conclut que le sommeil était la résultante d’une déafférentation sensorielle du cortex cérébral en accord avec la théorie passive du sommeil qui prévalait à l’époque. Une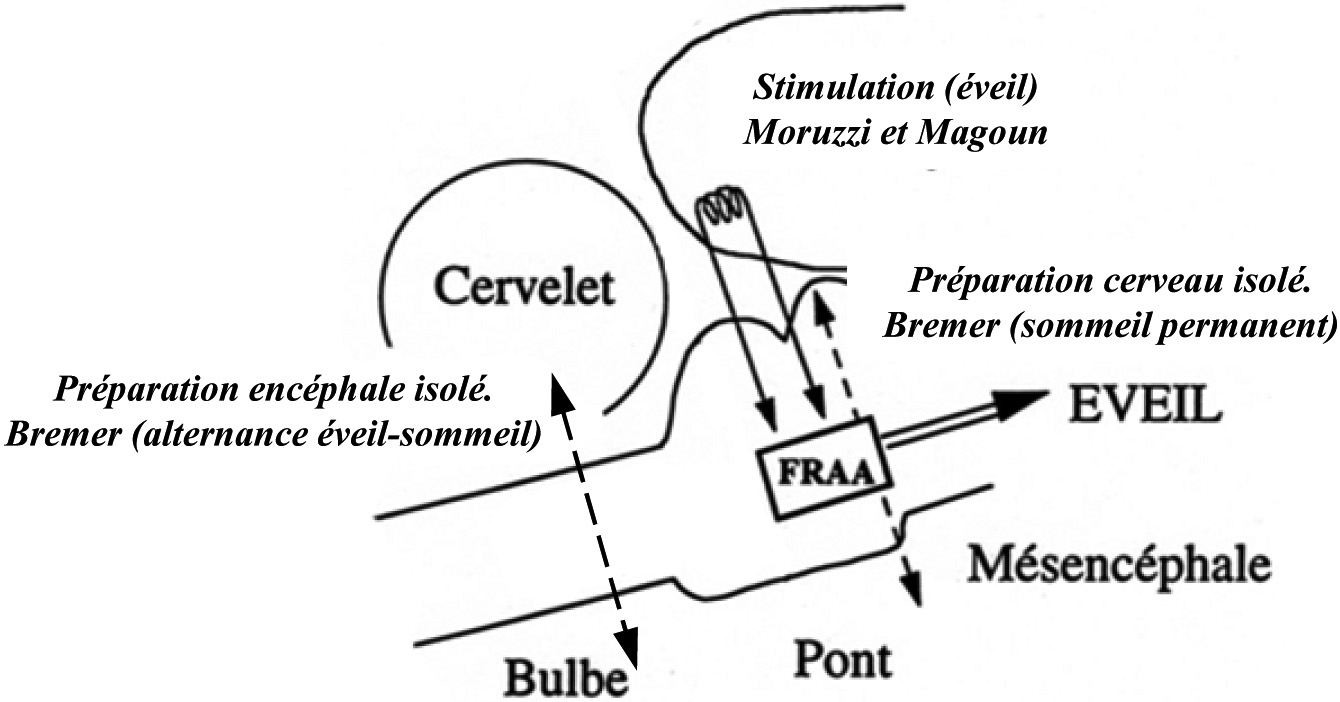
Fig. 2. — Importance du tronc cérébral dans l’activation corticale : le concept de formation réticulée activatrice ascendante (FRAA).
Les premières données expérimentales en faveur de l’importance du tronc cérébral dans l’éveil ont été rapportées par Bremer [4]. Elles montrent notamment que la déafférentation du mésencéphale produit un état de sommeil permanent dans la préparation dite du « cerveau isolé ». Moruzzi et Magoun ont confirmé ces résultats et ont montré que la stimulation électrique directe de la formation réticulée ponto-mésencéphalique provoque l’éveil chez le chat endormi [5].
quinzaine d’années plus tard, une nouvelle interprétation est formulée qui tient compte de la mise en évidence du système réticulaire activateur ascendant par
Moruzzi et Magoun [5]. C’est en effet dans la deuxième moitié des années 1940 que ces auteurs montrèrent que la stimulation électrique de la formation réticulée (FR), qui s’étend du bulbe rachidien jusqu’à la partie antérieure du mésencéphale, induisait une activation corticale (Figure 2). Ils en conclurent que le tronc cérébral contenait les structures responsables et nécessaires à l’activation corticale, une composante majeure de l’éveil.
Genèse de l’éveil : le système activateur ascendant
Aujourd’hui, il est bien établi que le système activateur ascendant comprend en réalité plusieurs ensembles neuronaux modulateurs dans le tronc cérébral, l’hypothalamus postérieur et le télencéphale basal dont la voie commune finale est le cortex céré- bral (Figure 3). Au niveau du tronc cérébral, il s’agit essentiellement des neurones sérotoninergiques des noyaux du raphé, noradrénergiques du locus cœruleus (LC), cholinergiques des noyaux du tegmentum pontique, dopaminergiques de la substance grise périaqueducale (PAG) ventrale et glutamatergiques de la FR. Les neurones de l’hypothalamus postérieur participant au maintien de l’éveil sont les neurones histaminergiques du noyau tubéro-mamillaire (TMN) et hypocrétinergiques des aires latérales (LH) et périfornicales. Enfin, les influences excitatrices en provenance du complexe du télencéphale basal impliquent essentiellement les neurones
Fig. 3. — Voies neuronales et neuromédiateurs dans le contrôle de l’éveil.
Coupe sagittale de cerveau de rongeur présentant les connexions anatomiques du système activateur ascendant impliqué dans l’activation corticale et le maintien de l’éveil.
L’activation corticale implique à la fois des projections directes (en jaune) et indirectes, celles-ci comportant un relais thalamique (en bleu).
Abréviations: Ach : Acétylcholine ; DA : Dopamine ; DRN : Noyau raphé dorsal ; FR : Formation réticulée ponto-mésencéphalique ; His : Histamine ; Hcrt : Hypocrétine ; LDTg : Noyau tegmental latérodorsal ; LC : Locus cœruleus ; LH : Hypothalamus latéral ; PPTg : Noyau tegmental pédonculopontin ; NA : Noradrénaline ; NBM : Noyau basal de Meynert ; TMN : Noyau tubéromamillaire ; vPAG: Substance grise périaqueducale ventrale ; 5-HT : sérotonine.
cholinergiques du noyau basal de Meynert. Ainsi, se définit un réseau exécutif de l’éveil qui regroupe essentiellement deux voies partant du tronc cérébral pour innerver le cortex de façon diffuse (Figure 3). Nous nous limiterons ici à une description des principales caractéristiques anatomiques et fonctionnelles des systèmes d’éveil au niveau du tronc cérébral et de l’hypothalamus.
Le système cholinergique des noyaux du tegmentum pontique [6, 7]
Ce système est majoritairement composé de deux groupes de neurones cholinergiques mésopontins distribués dans les noyaux tegmentaux latérodorsal (LDTg) et pédonculopontin (PPTg). Ces neurones, qui projettent sur les noyaux thalamiques, l’hypothalamus postérieur et le télencéphale basal, sont spécifiquement actifs pendant l’éveil et le sommeil paradoxal. Leur phénotype cholinergique a été confirmé par l’enregistrement extracellulaire des neurones du LDT combiné à des marquages juxtacellulaires chez l’animal en contention [8]. La plupart des neurones cholinergiques du LDTg et du PPTg participeraient ainsi au processus d’activation corticale, notamment via l’excitation directe des neurones thalamo-corticaux.
Le système noradrénergique du locus cœruleus (LC) [9]
Les neurones noradrénergiques du LC ont des projections ascendantes diffuses dans l’ensemble du cerveau comprenant l’hypothalamus, le télencéphale basal, le thalamus et le cortex cérébral. Ils présentent une décharge tonique et régulière pendant l’éveil, qui diminue pendant le sommeil lent et cesse durant le sommeil paradoxal.
Ce profil d’activité suggère l’implication du LC dans les processus d’éveil. Cette hypothèse est confortée par les propriétés éveillantes des antidépresseurs qui inhibent la recapture de noradrénaline et par le fait que la stimulation sélective des neurones noradrénergiques du LC dans une procédure d’optogénétique provoque des transitions immédiates du sommeil vers l’éveil et facilite l’éveil [10].
Ces données corroborent les résultats convergents de nombreuses études pharmacologiques. Ainsi, le blocage des récepteurs noradrénergiques de type alpha 2 induit de l’éveil probablement en levant le rétrocontrôle inhibiteur que la noradrénaline exerce sur ses propres neurones. Cependant, chez le rat, la destruction du LC n’a pas de conséquences sur l’éveil ou sur l’activation corticale. En réalité, des données récentes suggèrent que les neurones noradrénergiques du LC participent au maintien de l’éveil surtout dans des contextes comportementaux nécessitant une attention soutenue [11]. Ainsi, la lésion des neurones noradrénergiques du LC diminue la capacité des rats à rester éveillés lorsqu’ils sont placés dans un environnement nouveau et stimulant comprenant l’introduction de jouets et la présence d’un congénère. A contrario, les dérivés amphétaminiques et le modafinil, qui élèvent le tonus noradrénergique central, stimulent la vigilance aux dépens du sommeil [12].
Le système sérotoninergique des noyaux du raphé [13, 14]
Les corps cellulaires des neurones sérotoninergiques sont regroupés en neuf « noyaux » distincts dans le tronc cérébral. Parmi ces noyaux, le noyau raphe dorsal (DRN) contient la plus forte proportion de neurones sérotoninergiques et est à l’origine d’une innervation sérotoninergique dense dans le cerveau antérieur qui comprend l’hypothalamus, le télencéphale basal et le cortex cérébral. Le rôle de la sérotonine (5-hydroxytryptamine, 5-HT) dans la régulation des états de vigilance est sujet à controverse. Ainsi, les recherches initialement menées présentaient la 5-HT comme un neurotransmetteur facilitateur du sommeil lent. En effet, chez le chat, la destruction électrolytique des noyaux du raphé, de même que l’administration d’un inhibiteur de la synthèse de 5-HT (para-chlorophenylalanine ou pCPA), induisent une diminution de la 5-HT cérébrale associée à une insomnie totale et de longue durée [13]. Puis, d’autres approches expérimentales ont révélé une association étroite entre la 5-HT et l’éveil.
Ainsi, l’enregistrement de l’activité unitaire de neurones présumés sérotoninergiques du DRN chez le chat et le rat montre une décharge lente et régulière pendant l’éveil, qui diminue progressivement au cours du sommeil lent et cesse pendant le sommeil paradoxal. Actifs pendant l’éveil, les neurones sérotoninergiques exerceraient un contrôle inhibiteur sur le sommeil paradoxal [14]. De fait, les antidépres- seurs inhibiteurs sélectifs de la recapture de 5-HT (ISRS, comme la fluoxétine, la paroxétine, le citalopram, etc), qui élèvent le tonus sérotoninergique central, inhibent l’expression du sommeil paradoxal chez de nombreuses espèces, et en particulier la souris [15]. À l’opposé, l’invalidation génique ou le blocage pharmacologique des récepteurs de type 5-HT ou 5-HT provoque une hypersomnie en sommeil para1A 1B doxal [16, 17]. La sérotonine peut également moduler l’éveil et le sommeil lent en agissant sur les récepteurs de type 5-HT . Ainsi, les antagonistes des récepteurs 5-HT 2 2A induisent une augmentation du sommeil lent [18] et ont été proposés comme traitement contre l’insomnie. De même, le blocage des récepteurs 5-HT semble présenter 2C un intérêt pour promouvoir le sommeil lent [19], et il a été rapporté que le sommeil des patients déprimés est amélioré par des antidépresseurs ayant des propriétés antagonistes vis-à-vis de ces récepteurs, comme par exemple l’agomélatine [20].
À l’image des neurones noradrénergiques du LC, les neurones sérotoninergiques du raphé peuvent également être mis en jeu dans des circonstances particulières. En particulier, ils sont impliqués dans les modifications des états de vigilance après un stress aigu [21]. Par ailleurs, il a été suggéré qu’en intégrant des signaux d’urgence en provenance de la périphérie tels que l’hypercapnie, ces neurones induisent une réponse adé- quate pour la survie de l’animal : l’éveil [22]. Un exemple de cette mise en jeu pourrait être le réveil suivi d’une reprise de la respiration après un épisode d’apnée prolongée.
Le système dopaminergique de la substance grise périaqueducale ventrale (vPAG)
L’éveil est également associé à un tonus dopaminergique élevé [23]. Les effets éveillants et stimulants des composés comme la cocaïne ou les amphétamines sont liés au blocage de la recapture ou à la stimulation de la libération de dopamine (DA), des effets qui s’ajoutent à l’élévation du tonus noradrénergique déjà signalé plus haut. Les neurones dopaminergiques ascendants de l’aire tegmentale ventrale (VTA) étaient pressentis pour être responsables des effets éveillants de la DA.
Cependant, leur profil de décharge ne dépend pas des états de vigilance et leur lésion neurotoxique n’induit pas de réduction de l’éveil, mais provoque au contraire un état d’insomnie. Des études récentes suggèrent qu’en réalité, l’influence éveillante de la DA impliquerait des neurones localisés dans la vPAG [24], à proximité des neurones sérotoninergiques du DRN[25]. Ces neurones projettent essentiellement sur le LC, les neurones cholinergiques du télencéphale basal et le cortex préfrontal et leur lésion spécifique par micro-injection locale de la neurotoxine 6-hydroxydopamine (6-OH-DA) induit une augmentation des quantités de sommeil chez le rat. Ainsi, les neurones dopaminergiques de la vPAG, de par leurs projections sur les systèmes régulateurs du cycle éveil/sommeil, pourraient également apporter leur contribution au maintien de l’éveil.
Le système glutamatergique de la formation réticulée (FR) [26]
La FR est composée de neurones de grande taille et dispersés, et de nombreuses fibres. Elle s’étend sur tout le tronc cérébral et se divise en trois régions selon un axe caudo-rostral : médullaire ou bulbaire, pontique, et mésencéphalique. Majoritaire- ment, les projections ascendantes sont issues des neurones des noyaux réticulaires pontins caudal (PnC) et oral (PnO) et du noyau profond réticulaire mésencéphalique. Les neurotransmetteurs des populations neuronales hétérogènes de la FR n’ont pas encore été tous identifiés. Cependant, les phénotypes glutamatergique et GABAergique sont largement représentés parmi les neurones réticulaires. La plupart des neurones de la FR ponto-mésencéphalique ont une activité électrophysiologique tonique et rapide, et de niveau plus élevé pendant l’éveil et le sommeil paradoxal que pendant le sommeil lent [27]. De plus, la stimulation électrique de cette région induit une excitation des neurones thalamo-corticaux. Sur la base de ces résultats et de caractérisations neurochimiques, il est suggéré que le glutamate provenant de la FR ponto-mésencéphalique jouerait un rôle fondamental dans l’activation thalamo-corticale [26].
Le système histaminergique tubéro-mamillaire de l’hypothalamus [28]
Les neurones à histamine sont localisés dans le noyau tubéro-mamillaire de l’hypothalamus postérieur. Ils projettent de façon diffuse sur de nombreuses structures cérébrales associées à la régulation des états de vigilance telles que l’hypothalamus antérieur, le thalamus ou le cortex cérébral. L’importance des neurones à histamine dans l’éveil a initialement été révélée par les propriétés hypnotiques des composés antihistaminiques (antagonistes des récepteurs H1 de l’histamine), classiquement utilisés pour lutter contre les allergies. Ces données sont confortées par le profil d’activité électrophysiologique des neurones à histamine puisque ces derniers déchargent de manière tonique, spécifiquement pendant l’éveil [29]. De plus, les souris ne produisant pas d’histamine (souris knock-out dont le gène codant l’histidine décarboxylase, et donc la synthèse d’histamine, est invalidé) se caractérisent par une somnolence, ainsi qu’une incapacité à rester en veille dans diverses situations expérimentales telles que lors d’un changement de cage [30]. De par son rôle clé dans l’éveil, le système histaminergique est une cible d’intérêt pour le développement de nouvelles molécules favorisant la vigilance et l’attention. Ainsi, des antagonistes sélectifs des récepteurs histaminergiques de type H3 ont été développés pour contrer la somnolence dans les hypersomnies [31]. Ces molécules, en levant l’inhibition que l’histamine exerce sur ses propres neurones via les autorécepteurs H3, entraînent une facilitation de la neurotransmission histaminergique et, par conséquent, favorisent l’éveil.
Le système hypocrétinergique de l’hypothalamus latéral [32]
Les hypocrétines (ou orexines) sont deux neuropeptides, hcrt1 et hcrt2, issus du clivage d’un précurseur unique : la préprohypocrétine. Bien que la localisation des corps cellulaires des neurones à hypocrétine soit restreinte à l’hypothalamus latéral, ces neurones projettent dans de nombreuses structures cérébrales, dont certains noyaux associés aux cycles veille/sommeil [33]. Plusieurs arguments plaident en faveur d’un rôle facilitateur de l’hypocrétine dans l’éveil. Tout d’abord, l’administration intra-cérébroventriculaire d’hypocrétine prolonge l’état de veille en agissant notamment sur les neurones noradrénergiques du LC [34]. De plus, l’activité des neurones hypocrétinergiques est maximale pendant l’éveil et minimale au cours du sommeil [35, 36]. Enfin, leur activation in vivo , par la technique d’optogénétique, favorise les transitions du sommeil vers l’éveil [37].
En réalité, il est vite apparu que la transmission hypocrétinergique jouait un rôle clé dans une pathologie de l’éveil : la narcolepsie. De fait, une transmission hypocrétinergique altérée liée à l’absence du récepteur hypocrétinergique de type 2 dans un modèle canin ou à la dégénérescence des neurones à hypocrétine chez l’homme, s’accompagne des troubles comportementaux caractéristiques de la narcolepsie tels que la somnolence et/ou les attaques de cataplexie (chute brutale du tonus musculaire, sans perte de conscience). Ainsi, la découverte de ces neurones et la démonstration de leur implication majeure dans l’éveil permettent d’envisager de nouveaux traitements des troubles du sommeil [38]. D’ailleurs, les antagonistes des récepteurs à hypocrétine (bloquants mixtes des récepteurs de type 1 et de type 2) ont des propriétés hypnotiques chez l’homme. À l’inverse, le développement d’agonistes pourrait constituer une piste de grand intérêt pour lutter contre les hypersomnies dont la narcolepsie.
Au fil des ans, le rôle de ces différents ensembles dans la régulation de l’éveil a toutefois été remis en question. En effet, la lésion spécifique de certains de ces groupes, voire la lésion combinée de plusieurs d’entre eux, n’affecte, au mieux, que faiblement l’éveil. Il a alors été proposé que le maintien de l’éveil ne réside pas dans le bon fonctionnement d’un seul noyau mais requiert en réalité l’activité conjointe de plusieurs ensembles neuronaux. Cependant, ces données ne concernent que l’enregistrement du sommeil en conditions de « base », c’est-à-dire lorsque les animaux sont placés dans leur cage habituelle. Les données récentes sur les systèmes monoaminergiques et hypocrétinergique permettent de proposer une nouvelle signification à cette redondance : la complémentarité. Il n’existerait pas « un » éveil, mais « des » éveils qui, selon les situations comportementales, mettraient en jeu différents groupes neuronaux. À ce titre, l’étude des souris mutantes sans histamine ou sans hypocrétine s’avère riche d’enseignements. De fait, seules les souris sans hypocrétine montrent un déficit d’éveil face à un défi locomoteur [30]. Nous avons également mentionné précédemment le fait que l’impact de la lésion des neurones noradrénergiques du LC ne se révèle que dans un contexte comportemental nécessitant une attention soutenue [11]. Ce concept de complémentarité permet d’appréhender sous un angle nouveau la complexité des régulations mises en jeu pendant l’éveil.
Genèse du sommeil
Historiquement, le sommeil a d’abord été considéré comme un phénomène passif.
Ainsi, l’endormissement et la diminution de la perception sensorielle associée ne seraient que les conséquences d’une désactivation des systèmes d’éveil. En réalité, on sait aujourd’hui qu’il existe des processus actifs de genèse du sommeil mettant en jeu des réseaux neuronaux spécifiques.
Le système GABAergique de la zone ventrale de l’aire préoptique (VLPO) [39]
Les observations anatomopathologiques de Von Economo [2] et les expériences de lésions/stimulations [3] ont mis en évidence l’importance de l’hypothalamus anté- rieur dans la genèse du sommeil lent. Plus précisément, ce sont des lésions restreintes à la région préoptique de l’hypothalamus chez le chat qui provoquent une détérioration de l’architecture du cycle veille-sommeil avec une disparition parfois totale du sommeil lent. Le rôle prépondérant de la région préoptique dans l’endormissement a également été mis en évidence par des approches électrophysiologiques. Ainsi, les neurones de la partie latérale et surtout ventrolatérale (VLPO) de la région préoptique présentent une augmentation importante de leur fréquence de décharge juste avant ou en association avec la synchronisation corticale caractéristique du sommeil lent [40]. En outre, les neurones de la VLPO, qui contiennent en grande majorité des neuromédiateurs inhibiteurs (GABA et galanine), projettent massivement sur les systèmes d’éveil (TMN, DRN et LC), et reçoivent en retour de nombreuses afférences de ces noyaux. Ainsi, la majorité des neurones GABAergiques de la VLPO sont inhibés par la noradrénaline, l’acétylcholine et la sérotonine [41].
Le système GABAergique du cortex cérébral
Des études récentes suggèrent également la participation des interneurones GABAergiques du cortex cérébral dans la promotion du sommeil lent [42]. De fait, une petite proportion de ces neurones s’active pendant la période de récupération qui suit une privation de sommeil, en lien avec l’augmentation de la densité des ondes lentes corticales caractéristiques de cette période. Ces interneurones GABAergiques, qui se distinguent par l’expression de l’isoforme neuronale de l’oxyde nitrique synthétase (NOS), participeraient activement à l’homéostasie du sommeil lent. D’autres arguments, plus indirects, confortent cette hypothèse. D’une part, il existe des liens anatomiques entre ces neurones et ceux de l’éveil, cholinergiques et sérotoninergiques notamment, et d’autre part, l’oxyde nitrique (NO), classiquement associé au couplage neurovasculaire, pourrait également avoir des propriétés hypnogènes. Par ailleurs, une population d’interneurones GABAergiques produit un neuropeptide aux propriétés hypnogènes, la cortistatine (CST), qui présente une forte homologie avec la somatostatine (SST) [43]. Dans le système nerveux central, la présence de CST est restreinte à une sous-population d’interneurones GABAergiques de l’hippocampe et du cortex cérébral. Bien qu’appartenant à la même famille peptidergique, la CST se distingue de la SST par sa capacité à générer des réponses propres. Ainsi, l’administration intracérébroventriculaire de CST induit une augmentation des taux de sommeil lent profond chez des rats aussi bien en période d’activité qu’au repos [44]. De plus, la privation de sommeil total provoque une augmentation des taux tissulaires d’ARN messagers codant la CST, indiquant que la production de CST est positivement corrélée aux besoins en sommeil. L’ensemble de ces données suggère que la CST joue un rôle important dans l’homéostasie du sommeil. Ses récepteurs pourraient constituer de nouvelles cibles moléculaires pour une stratégie thérapeutique innovante de l’insomnie.
Bien qu’un rôle prépondérant soit attribué à la VLPO, la découverte récente de ces populations d’interneurones GABAergiques permet d’envisager l’existence de systèmes de régulation du sommeil lent plus complexes, à l’image des systèmes d’éveil, qui incluraient la participation non seulement de l’hypothalamus antérieur, mais aussi d’autres structures cérébrales telles que le cortex. Cependant, le manque de caractérisation anatomique et fonctionnelle de ces nouvelles populations ne permet pas, à l’heure actuelle, de les rattacher au modèle de régulation veille/sommeil.
La bascule veille-sommeil : le modèle d’interactions inhibitrices réciproques
C’est en se basant sur l’existence d’un contrôle inhibiteur réciproque entre les neurones du sommeil de la VLPO et certains neurones d’éveil, majoritairement monoaminergiques, qu’a été élaboré le modèle actuel de « bascule » entre systèmes d’éveil et de sommeil. Dans ce modèle (Figure 4), l’activité de l’un des systèmes inhibe celle de l’autre, désinhibant ainsi sa propre activité. Cette configuration a été proposée pour expliquer les transitions rapides éveil-sommeil. En effet, cette alternance inhibition/désinhibition, à l’image d’un interrupteur électrique (« flip-flop switch », [45]), évite la survenue de longs états transitionnels, intermédiaires, qui seraient dangereux pour la survie de l’animal puisque inadaptés. En particulier, un demi-éveil ne permettrait pas de faire face à des situations d’urgence (présence d’un prédateur, recherche de nourriture) et un demi-sommeil ne serait pas récupérateur.
Dans ce modèle, les neurones à hypocrétine, en établissant des relations asymétriques avec les systèmes d’éveil et de sommeil, se positionnent comme un levier stabilisant l’éveil. En effet, les neurones à hypocrétine envoient des projections excitatrices aux systèmes d’éveil du tronc cérébral et de l’hypothalamus, mais n’exercent pas d’influence inhibitrice directe sur les neurones de la VLPO, contrairement à ces derniers (Figure 4). Spécifiquement actifs pendant l’éveil, les neurones à hypocrétine stabiliseraient cet état de vigilance en renforçant l’activité des systè- mes d’éveil. Ainsi, les interactions inhibitrices entre les neurones du sommeil situés dans la VLPO et les neurones du système ascendant d’éveil régiraient les alternances veillle/sommeil, avec l’hypocrétine comme facteur stabilisateur (Figure 4). Ce modèle est conforté par les études cliniques sur la physiopathologie de la narcolepsie [46]. En effet, cette pathologie se caractérise par une dérégulation des transitions entre les différents états de vigilance, ce qui provoque leur instabilité et des difficultés à maintenir l’éveil (somnolence diurne) ou le sommeil (sommeil fragmenté). Or, chez l’homme, la narcolepsie serait causée par la dégénérescence sélective des neurones à hypocrétine. De plus, les symptômes de cette pathologie sont en accord avec les prédictions du modèle de « flip-flop switch » puisque, selon ce modèle, l’absence d’hypocrétine provoque des transitions anarchiques entre l’éveil et le sommeil.
CONCLUSION
L’identification des systèmes neuronaux responsables de la veille et du sommeil a permis de proposer un modèle de régulation des états de vigilance qui intègre
Fig. 4. — Voies neuronales et neuromédiateurs dans le contrôle de l’éveil.
Pendant le sommeil, les neurones de la VLPO sont actifs et inhibent les neurones d’éveil, monoaminergiques. De même manière, pendant l’éveil, ces derniers inhibent les neurones de la VLPO.
L’hypocrétine joue un rôle modulateur sur cet équilibre : les neurones qui libèrent ce neuropeptide sont actifs pendant l’(éveil et renforcent le tonus monoaminergique.
Abréviations : DA : Dopamine ; DRN : Noyau raphé dorsal ; GABA : Acide gamma amino butyrique ; Hcrt : Hypocrétine ; His : Histamine ; LC : Locus cœruleus ; LH : Hypothalamus latéral ; NA : Noradrénaline ; TMN : Noyau tubéro-mamillaire ; vPAG : Substance grise périaqueducale ventrale ; VLPO : Aire ventrolatérale préoptique ; 5-HT : Sérotonine.
également les mécanismes à l’origine de leur alternance. A cet égard, la découverte de l’hypocrétine et la démonstration de son implication dans la narcolepsie soulignent l’importance physiologique d’un contrôle efficace des transitions entre la veille et le sommeil. À l’heure où notre société malmène le sommeil (nous avons perdu une heure de sommeil en cinquante ans), il est nécessaire d’approfondir ces connaissances en étudiant, notamment, la capacité d’adaptation des systèmes de veille et du sommeil face à des perturbations de l’environnement. La découverte récente des liens entre réduction du temps de sommeil, surpoids et pathologies cardiovasculaires en fait plus que jamais un enjeu de santé publique.
BIBLIOGRAPHIE [1] Ohayon M.M. — Prévalence et comorbidité des troubles du sommeil dans la population générale. La revue du Praticien , 2007, 57 , 1521-1528.
[2] Von Economo M. — Sleep as a problem of localization.
J. Nerv. Mental Disease , 1931, 71 , 249-269.
[3] Nauta W.H.J. — Hypothalamic regulation of sleep in rats. Experimental study. J. Neurophy- siol., 1946, 9 , 285-316.
[4] Bremer F. — Cerveau isolé et physiologie du sommeil.
Comptes Rendus Société de Biologie, 1935, 118 , 1235-1241.
[5] Moruzzi G., Magoun H.W. — Brain stem reticular formation and activation of the EEG.
Electroencephalogr. Clin. Neurophysiol., 1949, 1 , 455-473.
[6] Steriade M. — Acetycholine systems and rhythmic activities during the waking-sleep cycle.
Prog. Brain Res., 2004, 145 , 179-196.
[7] Jones B.E. — Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Ann. N Y Acad. Sci. , 2008, 1129 , 26-34.
[8] Boucetta S., Jones B.E. — Activity profiles of cholinergic and intermingled GABAergic and putative glutamatergic neurons in the pontomesencephalic tegmentum of urethaneanesthetized rats. J. Neurosci. , 2009, 29 , 4664-4674.
[9] Berridge C.W., Waterhouse B.D. — The locus coeruleus-noradrenergic system : modulation of behavioral state and state-dependent cognitive processes. Brain Res. Brain Res. Rev ., 2003, 42 , 33-84.
[10] Carter M.E., Yizhar O., Chikahisa S. et al. — Tuning arousal with optogenetic modulation of locus coeruleus neurons.
Nat. Neurosci ., 2010, 13 , 1526-1533.
[11] Gompf H.S., Mathai C., Fuller P.M. et al. — Locus ceruleus and anterior cingulate cortex sustain wakefulness in a novel environment.
J. Neurosci., 2010, 30 , 14543-14551.
[12] Mitchell H.A., Weinshenker D. — Good night and good luck : norepinephrine in sleep pharmacology. Biochem. Pharmacol., 2010, 79 , 801-809.
[13] Jouvet M. — Sleep and serotonin : an unfinished story.
Neuropsychopharmacology, 1999, 21 , 24S-27S.
[14] Ursin R. — Serotonin and sleep. Sleep Med. Rev ., 2002, 6 , 55-69.
[15] Monaca C., Boutrel B., Hen R. et al. — 5-HT1A/1B receptor-mediated effects of the selective serotonin reuptake inhibitor, citalopram, on sleep : studies in 5-HT1A and 5-HT1B knockout mice. Neuropsychopharmacology, 2003, 28 , 850-856.
[16] Boutrel B., Franc B., Hen R. et al. — Key role of 5-HT1B receptors in the regulation of paradoxical sleep as evidenced in 5-HT18 knock-out mice.
J. Neurosci., 1999, 19 , 3204-3212.
[17] Boutrel B., Monaca C., Hen R. et al. — Involvement of 5-HT1A receptors in homeostatic and stress-induced adaptive regulations of paradoxical sleep : studies in 5-HT1A knock-out mice. J. Neurosci., 2002, 22 , 4686-4692.
[18] Popa D., Lena C., Fabre V. et al. — Contribution of 5-HT2 receptor subtypes to sleepwakefulness and respiratory control, and functional adaptations in knock-out mice lacking 5-HT2A receptors. J. Neurosci., 2005, 25 , 11231-11238.
[19] Sharpley A.L., Elliott J.L., Attenburrow M.J., Cowen P.J. — Slow wave sleep in humans :
role of 5-HT2A and 5-HT2C receptors. Neuropharmacology, 1994, 33 , 467-471.
[20] Kasper S, Hamon M. — Beyond the monoaminergic hypothesis : agomelatine, a new antidepressant with an innovative mechanism of action. World J. Biol. Psychiatry, 2009, 10 , 117-126.
[21] Rachalski A., Alexandre C., Bernard J.F. et al. — Altered sleep homeostasis after restraint stress in 5-HTT-/-knock-out male mice: a role for hypocretins.
J. Neurosci., 2009, 29 , 15575- 15585.
[22] Buchanan G.F., Richerson G.B. — Central serotonin neurons are required for arousal to CO2. Proc. Natl. Acad. Sci. U S A, 2010, 107 , 16354-16359.
[23] Monti J.M., Monti D. — The involvement of dopamine in the modulation of sleep and waking. Sleep Med. Rev. , 2007, 11 , 113-133.
[24] Lu J., Jhou T.C., Saper C.B. — Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J. Neurosci ., 2006 , 26 , 193-202.
[25] Bonnavion P., Bernard J.F., Hamon M. et al. — Heterogeneous distribution of the serotonin 5-HT receptor mRNA in chemically identified neurons of the mouse rostral brainstem:
1A Implications for the role of serotonin in the regulation of wakefulness and REM sleep. J. Comp.
Neurol., 2010 , 518 , 2744-2770.
[26] Jones B.E. — Arousal systems.
Front Biosci. , 2003, 8 , s438-451.
[27] Steriade M. — Mechanisms underlying cortical activation : neuronal organization and properties of the midbrain reticular core and intralaminar thalamic nuclei. In Brain mechanisms of perceptual awareness and purposeful behavior (Pompeiano O, Ajmone-Marsan C, editors. New
York : Raven Press), 1981, 327-377.
[28] Lin J.S., Anaclet C., Sergeeva O.A. et al. — The waking brain : an update. Cell. Mol. Life Sci., 2011, 68 , 2499-2512.
[29] Takahashi K., Lin J.S., Sakai K. — Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J. Neurosci. , 2006, 26 , 10292-10298.
[30] Anaclet C., Parmentier R., Ouk K. et al. — Orexin/hypocretin and histamine : distinct roles in the control of wakefulness demonstrated using knock-out mouse models.
J. Neurosci. , 2009, 29 , 14423-14438.
[31] Schwartz J.C. — The histamine H3 receptor : from discovery to clinical trials with pitolisant.
Br. J. Pharmacol., 2011, 163 , 713-721.
[32] Bonnavion P., De Lecea L. — Hypocretins in the control of sleep and wakefulness.
Curr.
Neurol. Neurosci. Rep., 2010, 10 , 174-179.
[33] Peyron C., Tighe D.K., Van Den Pol A.N. et al. — Neurons containing hypocretin (orexin) project to multiple neuronal systems.
J. Neurosci., 1999, 18 , 9996-10015.
[34] Bourgin P., Huitron-Redendiz S., Spier A.D. et al. — Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons.
J. Neurosci., 2000, 20 , 7760-7765.
[35] Lee M.G., Hassani O.K., Jones B.E. — Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J. Neurosci., 2005, 25 , 6716-6720.
[36] Mileykovskiy B.Y., Kiyashchenko L.I., Siegel J.M. — Behavioral correlates of activity in identified hypocretin/orexin neurons, Neuron ., 2005 , 46 , 787-798.
[37] Adamantidis A.R., Zhang F., Aravanis A.M. et al. — Neural substrates of awakening probed with optogenetic control of hypocretin neurons.
Nature , 2007, 450 , 420-424.
[38] Scammell T.E., Winrow C.J. — Orexin receptors : pharmacology and therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. , 2011, 51 , 243-266.
[39] Luppi P.H. — Neurochemical aspects of sleep regulation with specific focus on slow-wave sleep.
World J. Biol. Psychiatr., 2010, 11 , 4-8.
[40] Szymusiak R., Alam N., Steininger T.L. et al. — Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats.
Brain Res. 1998, 803 , 178-188.
[41] Gallopin T., Fort P., Eggermann E. et al. — Identification of sleep-promoting neurons in vitro . Nature, 2000, 404 , 992-995.
[42] Kilduff T.S., Cauli B., Gerashchenko D. — Activation of cortical interneurons during sleep:
an anatomical link to homeostatic sleep regulation ? Trends Neurosci. , 2011, 34 , 10-19.
[43] De Lecea L., Criado J.R., Prospero-Garcia O. et al. — A cortical neuropeptide with neuronal depressant and sleep-modulating properties.
Nature, 1996, 381 , 242-245.
[44] Bourgin P, Fabre V, Huitron-Resendiz S. et al. — Cortistatin promotes and negatively correlates with slow-wave sleep.
Eur. J. Neurosci., 2007, 26, 729-738.
[45] Saper C.B., Fuller P.M., Pedersen N.P. et al. — Sleep state switching. Neuron , 2010, 68 , 1023-1042.
[46] Dauvilliers Y., Arnulf I., Mignot E. — Narcolepsy with cataplexy. Lancet , 2007, 369 , 499-511.
DISCUSSION
M. Henri-Philippe HUSSON
Les anti-histaminiques de type H1 sont connus depuis longtemps pour leurs propriétés hypnotiques. Est-ce encore une voie intéressante versus les anti-H3 ?
Les composés anti-histaminergiques antagonistes des récepteurs H1 (diphenhydramine, doxylamine, et chlorpheniramine) de première génération ont effectivement été utilisés comme hypnotiques dans les insomnies légères. Cependant, ces composés présentent également des propriétés antagonistes vis-à-vis des récepteurs muscariniques contribuant à des effets indésirables. Leur demi-vie longue semble également responsable d’effets prolongés, donc indésirables, le jour qui suit la prise de ces composés. De ce fait, et aussi parce qu’une nouvelle génération d’hypnotiques visant le système GABAergique est aujourd’hui disponible, ces composés sont peu prescrits. Cependant, le système histaminergique reste une cible majeure pour le développement de nouveaux composés pharmacologiques visant cette fois-ci à améliorer la qualité de l’éveil et à lutter contre la somnolence, tels que les antagonistes des récepteurs H3.
* Neurotransmetteurs et sommeil : UMR975 Cricm — Inserm/CNRS/UPMC, Faculté de médecine Pitié- Salpêtrière — 91 bld de l’Hôpital — 75013 Paris, e-mail : veronique.fabre@upmc.fr ** Membre correspondant de l’Académie nationale de médecine, e-mail : michel.hamon@upmc.fr Tirés à part : Docteur Véronique Fabre, même adresse Article reçu le 23 septembre 2011, accepté le 10 octobre 2011
Bull. Acad. Natle Méd., 2011, 195, no 7, 1551-1565, séance du 11 octobre 2011