
Summary
Perinatal brain damage following hypoxia-ischemia has long been considered irreversible, but recent rodent studies show that various insults can induce de novo neurogenesis in the adult brain. Here we examined whether acute hypoxia could trigger neurogenesis in the developing rat brain. In vitro , we examined the impact of transient hypoxia on cultured embryonic rat neurons. In vivo , we monitored the time course of brain damage in the CA1 layer of the hippocampus of one-day-old rats after exposure to hypoxia. The extent of cell loss and regeneration was evaluated after staining with DAPI. Newly generated cells were characterized in the subventricular zone by immunohistochemistry, 20 days post-exposure. The viability of cultured neurons was reduced by 36 % when measured 96 h after 6 h of hypoxia, and a significant number of cell nuclei showed apoptotic features. In contrast, cell numbers increased by 14 % 96 h after 3 h of hypoxia. The Bax/Bcl 2 ratio tended to increase after 6 h of hypoxia and to decrease after 3 h of hypoxia.In the CA1 subfield of the hippocampus, hypoxia reduced the total number of cells by 27 % on day 6-7 postreoxygenation, and histopathological hallmarks of apoptosis were observed. This cell deficit was followed by gradual recovery, starting on day 20, suggesting a repair mechanism. BrdU incorporation in the subventricular zone showed an accumulation of proliferating cells expressing the neuronal marker NeuroD. These data show that post-hypoxic neurogenesis can occur during development.
Si les progrès de la médecine périnatale ont réduit l’incidence des encéphalopathies hypoxiques-ischémiques chez le nouveau-né à terme, elles n’ont cependant pas disparu, et ces accidents demeurent fréquents chez les enfants prématurés. Longtemps les lésions cérébrales post-asphyxiques ont été considérées comme irréversibles, responsables soit de décès, soit de séquelles motrices, cognitives ou épileptiques durables, incitant le clinicien a une attitude fataliste [1-3].
De nombreuses études récentes ont décrit en détail les séquences physiopathologiques conduisant à la mort neuronale soit par nécrose, soit par les mécanismes de la mort programmée ou apoptose [4-6]. Après une agression hypoxique, le devenir des cellules nerveuses peut dépendre d’un déséquilibre entre l’expression de protéines pro-apoptotiques comme Bax ou anti-apoptotiques, donc protectrices, comme Bcl-2. En écho à des observations déjà anciennes suggérant que le cerveau immature serait plus résistant à l’hypoxie que celui de l’adulte [7-8] et que la plasticité cérébrale est plus efficace dans les périodes initiales du développement [9-11], des études réalisées dans divers modèles montrent que des épisodes hypoxiques ou des convulsions peuvent induire une neurogenèse dans certains sites du cerveau adulte comme la zone sous-ventriculaire ou le gyrus denté de l’hippocampe [12-13].
Les travaux présentés ici avaient pour but d’élucider le devenir de neurones du cerveau en développement après exposition transitoire à une hypoxie, avec pour hypothèse que cette dernière était susceptible d’induire une neurogenèse potentiellement « réparatrice ».
Dans cette perspective, deux séries d’expérimentations ont été programmées in vitro et in vivo [14]. In vitro , des neurones embryonnaires de rat en culture ont été exposés à des périodes d’hypoxie de durée différente, et les mécanismes impliqués dans leur devenir ont été analysés. In vivo , après exposition de rats nouveau-nés à une hypoxie supposée reproduire, approximativement, une asphyxie périnatale, l’évolution des neurones a été observée au niveau de la couche des cellules pyramidales du champ CA1 de l’hippocampe, particulièrement sensible à la privation d’oxygène.
Matériels et Méthodes
Études in vitro sur neurones en culture.
Une culture primaire de neurones a été obtenue à partir du cerveau antérieur d’embryons de rat de quatorze jours. Ces cellules ont été cultivées en conditions standard, à 37° C dans une atmosphère humidifiée composée de 95 % d’air et de 5 % de CO2. Après six jours, elles ont été incubées en présence d’un mélange de 95 % d’azote et 5 % de CO2 pendant trois ou six heures, enfin remises dans les conditions initiales. Les cellules témoins ont été conservées en normoxie.
La morphologie cellulaire a été observée en microscopie à contraste de phase, la pureté de la culture de neurones a été évaluée grâce à des anticorps dirigées contre l’Enolase Spécifique des Neurones (NSE) et la « glial fibrillary acidic protein » (GFAP) marqueur des astrocytes. La viabilité cellulaire a été mesurée par la méthode du bleu Trypan et par spectrophotométrie en utilisant le méthyltetrazolium ou MTT [14].
Les aspects morphologiques de l’apoptose, la nécrose et les mitoses ont été visualisés après fixation des cellules et coloration des noyaux par le 4,6—diamidino-2 phenylindole (DAPI). L’apoptose a été également évaluée par l’étude de l’expression de protéines régulatrices comme Bax, Bcl-2, caspase-3 ou p. 53 [16-18].
Études in vivo sur rats nouveau-nés.
Des rats de moins de vingt-quatre heures ont été partagés en deux groupes, les uns placés durant vingt minutes dans une chambre thermostatée à 36°, ventilée avec 100 % d’azote, les autres pendant le même temps à 21 % d’O2 et 79 % de N2.
Tous les jeunes rats étaient ensuite replacés avec leur mère. Dans ces conditions, la mortalilé des rats hypoxiques était de 4 % ; chaque portée était ensuite ramenée à dix sujets, cinq témoins et cinq hypoxiques. Des prélèvements sanguins ont été obtenus par décapitation immédiatement après exposition à l’hypoxie ainsi que chez les témoins pour mesurer pH, pO2 et pCO2.
L’évaluation de la perte cellulaire post-hypoxique a été réalisée à différents temps sur des coupes de cerveau de 20 µm prélevées au niveau de la partie antérieure de l’hippocampe. Les coupes ont été fixées et colorées avec de la thionine et les analyses morphologiques ont été faites par microscopie couplée à un analyseur d’images
numérique. Des coupes de cerveau adjacentes ont été mises en présence de DAPI pour visualiser les noyaux et permettre un comptage de la densité cellulaire et la caractérisation de l’apoptose et de la nécrose. Dans ces conditions, les cellules nécrosées présentent, à l’examen microscopique, un petit noyau réfringent dont la chromatine est uniformément dispersée ; l’apoptose est, quant à elle, reflétée par une chromatine condensée et fragmentée aisément reconnaissable. Par ailleurs, l’expression des protéines régulatrices de l’apoptose a été étudiée par immuno-histo-chimie [19, 20] Pour certaines phases de l’expérimentation, la prolifération cellulaire a été mesurée en fonction du temps après l’épisode hypoxique dans la zone sous-ventriculaire, site neurogénique chez le rongeur adulte [21]. Pour ce faire, les rats ont reçu une injection quotidienne de bromo-désoxy-Uridine (BrdU, 50mg/Kg, iP) pendant neuf jours précédant le sacrifice. La BrdU était objectivée sur des coupes cérébrales par immo-histo-chimie. Des marqueurs spécifiques étaient utilisés pour identifier le phénotype des nouvelles cellules [22].
RÉSULTATS
Effets de l’hypoxie in vitro
L’exposition à l’azote a réduit la pO2 du milieu de culture à 20 % de sa valeur initiale. Après une hypoxie de six heures, des altérations neuronales ont pu être observées dès 48 heures après réoxygénation et le nombre des cellules survivantes était réduit de 36 % à 96 heures (Fig .1) [23-24].
A 48 heures, on pouvait identifier un nombre de noyaux apoptotiques significativement plus élevé que celui des noyaux nécrotiques.
Après trois heures d’hypoxie, au contraire, on ne constatait aucun dommage neuronal apparent et le nombre de neurones était augmenté de 14 % (P < 0,01) à 96 heures (Fig.1), sans augmentation du nombre de cellules gliales.
Les analyses immuno-histochimiques ont montré qu’à 48 heures, et encore plus à 96 heures, une hypoxie de six heures s’accompagne d’une élévation de rapport Bax/Bcl-2 du fait d’une surexpression de Bax évocatrice d’un processus d’apoptose.
A l’opposé, après trois heures d’hypoxie, une augmentation de l’expression de la protéine anti-apoptotique Bcl-2 fait baisser le rapport Bax/Bcl-2 (fig. 2), tandis que les neurones expriment certaines protéines associées à la division cellulaire comme le « Proliferative cell nuclear antigen » [15].
Effets de l’hypoxie in vivo
Les rats nouveau-nés exposés à une hypoxie de vingt minutes présentaient dans le sang mêlé une profonde baisse de PO2 et du pH et une forte hypercapnie (tableau), ces valeurs étant plus altérées que ce qu’on constate en clinique humaine.
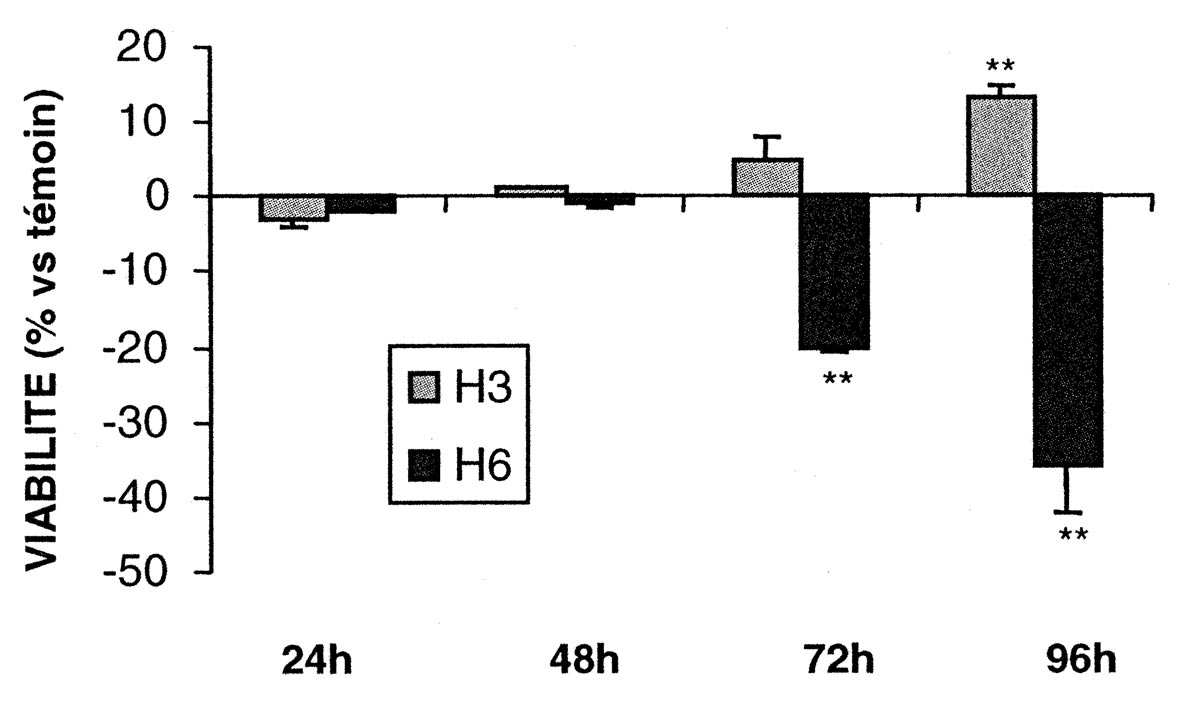
Témoins Hypoxie P< pO2 60,7 fi 7,4 11,8 fi 4,8 0,01 mmHg pCO2 35,6 fi 7,3 130,5 fi 6,9 0,01 mmHg pH 7,40 fi 0,03 6,62 fi 0,03 0,01 Fig. 1. — Évolution de la viabilité cellulaire de neurones en culture exposés à une hypoxie soit de six heures (H6) soit de trois heures (H3) par comparaison avec des cultures témoins maintenues en normoxie. Résultats obtenus sur cinq échantillons différents et exprimés en moyennes fi une déviation standard (DS). Différences avec les témoins statistiquement significatives : ** p<0,01 (Test de Dunnett pour comparaisons multiples).
Les rats hypoxiques ont présenté par la suite une réduction durable de la croissance corporelle et une altération transitoire du poids cérébral [19]. Alors que les pathologies post-asphyxiques de la substance blanche sont fréquentes, à l’origine de la leucomalacie périventriculaire observée chez les grands prématurés [25], nous n’avons pas mis en évidence, dans nos conditions expérimentales, de lésions décelables dans la substance blanche. En revanche, des lésions neuronales ont touché différentes régions du cerveau, comme le cortex cérébral et l’hippocampe et elles étaient particulièrement notables au niveau des cellules pyramidales du champ CA1 de l’hippocampe. La figure 3, partie supérieure, montre que dans la couche CA1 la densité cellulaire décline progressivement, son altération étant maximale six à sept jours après la réoxygénation avec une perte de 27 % par comparaison aux témoins.
L’histopathologie a permis de confirmer l’accumulation de neurones avec une condensation nucléaire et une fragmentation de la chromatine caractéristique du processus apoptotique qui a été par la suite confirmé par l’utilisation d’un anticorps spécifique nouvellement disponible (Apostain F7-26, AbCys SA, Paris,).
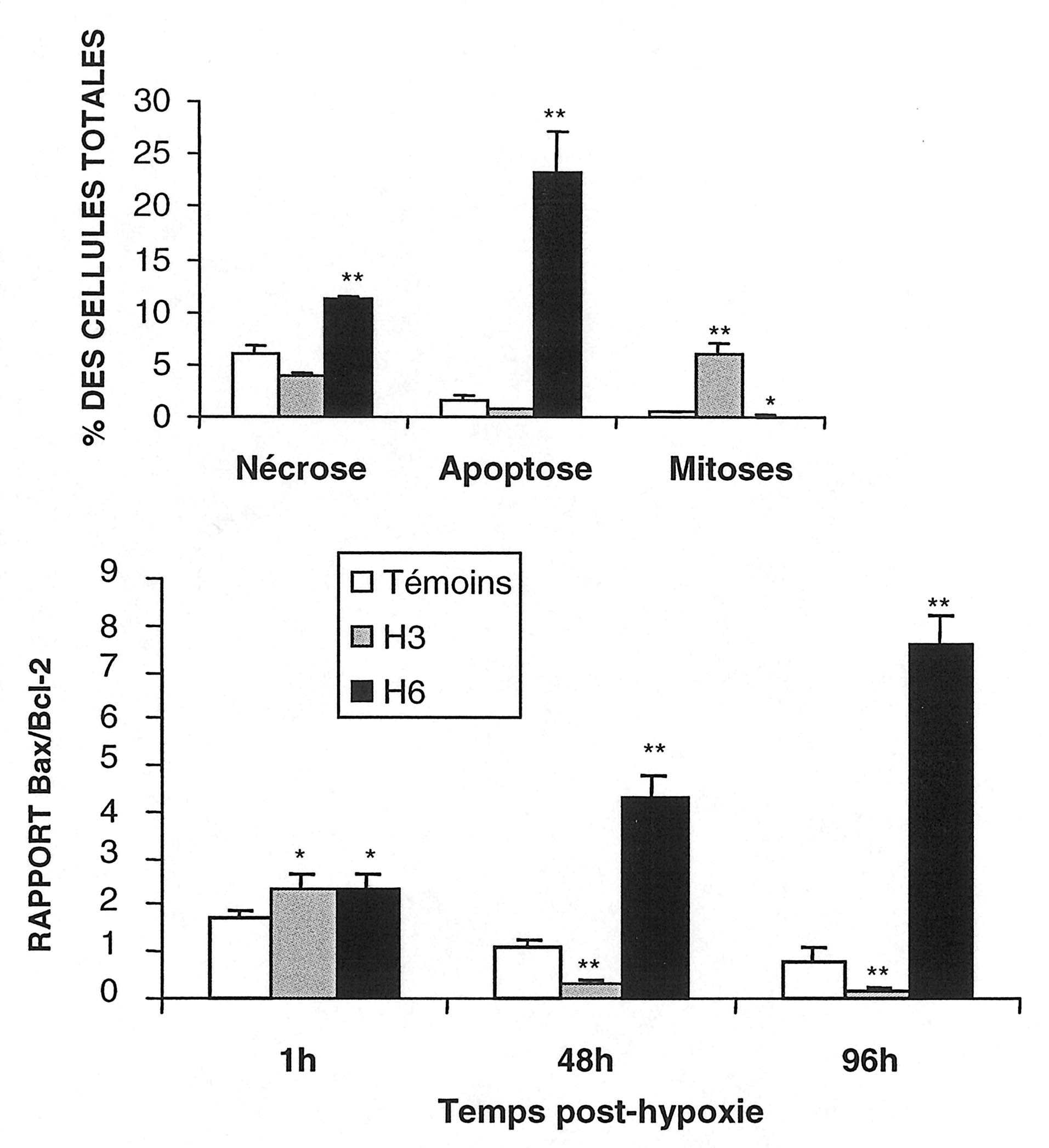
FIG. 2. —
Panneau supérieur — Proportions de nécroses, d’apoptoses et de mitoses dans les cultures de neurones 96 heures après l’hypoxie. Les caractéristiques morphologiques ont été comptabilisées dans les différents groupes de cultures après incorporation nucléaire de DAPI. Résultats obtenus sur au moins 5 échantillons et exprimés en moyennes fi DS. Différences avec les témoins : *, P<0,05 ; **, P<0,01 (Test de Dunnett).
Panneau inférieur — Evolution du rapport des protéines Bax/Bcl2 dans les neurones en culture exposés à une hypoxie de six heures (H6) ou de trois heures (H3) et dans les cultures témoins. Les mesures histo-chimique ont été effectuées une heure après le début de l’hypoxie puis 48 et 96 heures après ré-oxygénation, ainsi que dans les cultures témoins. L’intensité de la fluorescence a été calculée au moyen d’un logiciel photoscope Adobe et le rapport entre les taux de protéines était établi à chaque temps d’examen. Les résultats sont des moyennes fi SD calculés à partir de trois échantillons différents : *P<0,05 ; **P<0,01 (Test de Dunnett).
L’immuno-histologie et le comptage cellulaire ont montré que ce sont les cellules NSE positives, donc les neurones, qui étaient principalement affectées. Après une surexpression transitoire de Bcl-2 à trois jours, on assiste par la suite à une élévation marquée de Bax qui est maximale à treize jours (Fig.3). Cependant, un suivi plus prolongé a montré que le déficit cellulaire s’atténuait ensuite progressivement avec une restitution numérique à partir du vingtième jour (Fig. 3), ce qui suggère un processus de réparation.
L’examen des coupes de cerveau prélevées à vingt jours après l’injection de BrdU a révélé une prolifération de cellules dans la zone sous-ventriculaire des rats exposés à une hypoxie (Fig. 4). Ces cellules exprimaient la neuro D, marqueur de neurones immatures, et tendaient à migrer en cordon vers l’hippocampe le long du faisceau périventriculaire postérieur.
Discussion
Nos deux types d’expérimentation montrent un même processus de division cellulaire à la suite d’un épisode hypoxique, tant in vitro sur neurones embryonnaires que in vivo chez le rat nouveau-né. In vitro après six heures d’hypoxie, une forte proportion de neurones meurt par apoptose. De même in vivo c’est par apoptose que se fait la plus grande part de la perte des neurones pyramidaux dans le champ CA1 de l’hippocampe. Il est maintenant bien établi que ce phénomène reproduisant une partie du programme de mort cellulaire du développement normal, constitue l’évé- nement crucial de la constitution de lésions neuronales retardées sévères après hypoxie [26-29]. En plus d’aspects morphologiques caractéristiques, l’apoptose se traduit, dans nos modèles, par une expression séquentielle de protéines régulatrices spécifiques. En effet, contrairement à la nécrose qui est un processus passif, l’apoptose est caractérisée par l’induction de gènes spécifiques, et c’est la balance entre l’expression de gènes pro-apoptotiques comme Bax, et anti-apoptotiques comme Bcl-2 qui détermine le devenir des neurones exposés à l’hypoxie [15-29].
Lorsqu’on expose les cultures de neurones à une hypoxie « non létale » de trois heures, on assiste à une surexpression de protéines protectrices comme Bcl-2 qui favorisent la survie cellulaire. L’apoptose constitue une sorte d’avortement de la reprise du cycle de division cellulaire, un processus anormal ou conflictuel entre activation et arrêt de ce cycle [30, 31]. Ceci concorde avec la démonstration qu’un inhibiteur du cycle de division cellulaire protège les neurones des lésions induites par l’hypoxie [15-24].
Nos observations suggèrent qu’une hypoxie de durée modérée est susceptible d’induire une prolifération neuronale en provoquant l’expression de protéines associées à la division cellulaire comme le « Proliferating cell nuclear antigen » (PCNA) ou de survie comme Bcl-2.
Chez le rat nouveau-né, l’observation d’une neurogenèse consécutive à l’hypoxie dans la zone germinative sous-ventriculaire confirme des travaux sur l’animal adulte ou la genèse de nouveaux neurones apparaît non seulement comme un mécanisme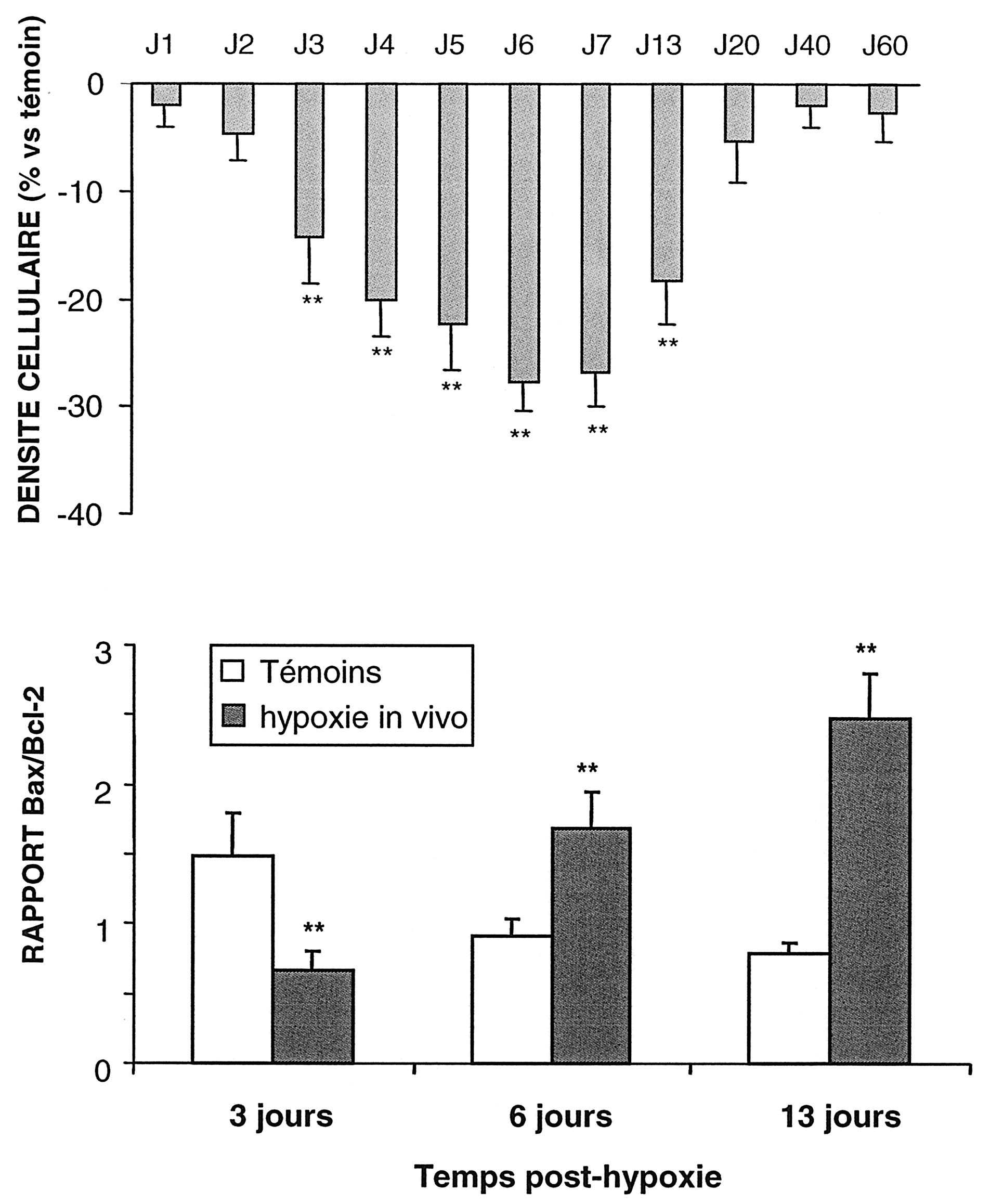
FIG. 3. —
Panneau supérieur — Influence d’une hypoxie de vingt minutes in vivo sur la densité cellulaire dans la couche CA1 de l’hippocampe de rat nouveau-né.
Aux différents temps le nombre total de cellules par mm3 était compté au moyen d’un réseau oculaire de 1/400mm2 après coloration des noyaux par le DAPI chez les rats hypoxiques et chez les témoins.
Différences significatives Vs témoins : **, P<0,01 (Test de Dunnett) Panneau inférieur — Evolution du rapport des protéines Bax/Bcl2 dans la couche cellulaire CA1 de rats exposés à une hypoxie néonatale et de rats témoins. Les mesures immuno-histochimiques ont été faites à trois, six et treize jours après hypoxie et chez les témoins. Résultats exprimés en moyennes (fiDS) sur trois échantillons différents. Différences significatives : **, P<0,01 (test de Dunnett).
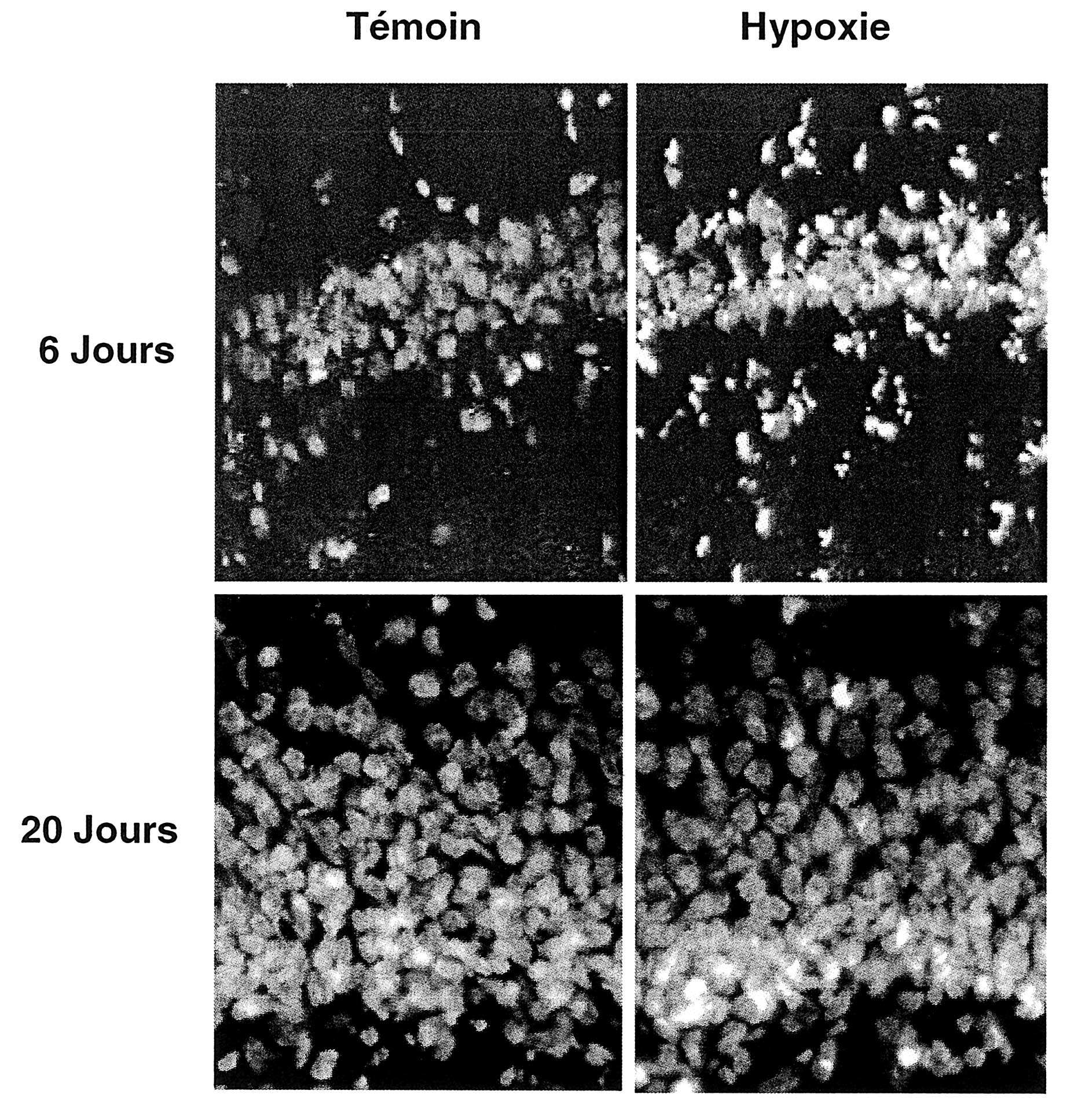
FIG. 4. — Évolution de la densité cellulaire dans la couche CA1 de l’hippocampe six et vingt jours après hypoxie chez des rats nouveau-nés et leurs témoins. Coloration des noyaux par DAPI (grossissement x40) un profil similaire était observé chez trois sujets.
transitoire de réparation, mais aussi comme un phénomène continu, la vie durant [32, 33]. Ces nouveaux neurones peuvent migrer et s’intégrer à des réseaux dans des sites spécifiques [34]. Une neurogenèse comparable a aussi été décrite dans le cortex cérébral [35]. L’observation d’une neurogenèse après une phase d’apoptose suggère une relation entre ces deux processus. Cependant, il est important de souligner que dans une étude plus récente, il a été possible d’induire une neurogenèse après cinq minutes d’hypoxie chez le rat nouveau-né sans apoptose apparente. Les cellules nouvellement générées ont présenté transitoirement un phénotype glial et ont révélé des propriétés fonctionnelles [36, 37]. Ceci suggère que certaines conditions environnementales peuvent stimuler les capacités neurogéniques du cerveau, en parti-
culier du cerveau en développement, en dehors de toute atteinte lésionnelle, et donc sans composante réparatrice.
Les mécanismes impliqués dans la neurogenèse postnatale font intervenir des facteurs neurotrophiques comme IGF1, EGF, FGF2, NGF ou l’érythropoïétine mais on ignore toujours le facteur déclenchant. Les stéroïdes surrénaliens sont impliqués dans la régulation de la neurogenèse au niveau du gyrus denté du cerveau adulte par l’intermédiaire des récepteurs N-methyl-D-aspartate (NMDA) [38]. Ceci pourrait expliquer l’effet néfaste du stress pré-ou postnatal sur les capacités d’apprentissage, allant de pair avec une inhibition de la neurogenèse dans l’hippocampe tant chez le rat que chez le singe [39, 40].
Le devenir des neurones néoformés n’est pas totalement élucidé. Il a été montré qu’ils persistent dans l’hippocampe de rongeurs adultes et que, chez le rat en développement, ils expriment la protéine synaptique synapsine I, témoignant de leur fonctionnalité. Par ailleurs, une incertitude demeure considérant que si ces neurones ne s’intégraient pas de façon adéquate dans les structures cérébrales ils pourraient être cause de réseaux aberrants comme dans l’hétérotopie de la fœtopathie alcoolique [41]. La neurogenèse post-hypoxique pourrait constituer un phénomène de protection impliquant des mécanismes possiblement différents de ceux qui interviennent dans l’induction d’une tolérance par un préconditionnement hypoxique bref [42], ou dans l’effet préventif des lésions neuronales par hypothermie [43].
La neurogenèse pourrait être considérée comme un phénomène de plasticité céré- brale à rapprocher de l’hypertrophie d’une région cérébrale controlatérale après hémisphérectomie chez le rat [44].
Nos résultats ajoutés à des données comparables obtenues dans d’autres modèles expérimentaux, par différents auteurs, amènent à contredire le dogme fondamental admis pendant des décennies, selon lequel le système nerveux central ne pourrait jamais être le siège de divisions neuronales passée la période fœtale ou néonatale précoce.
Reste à s’interroger sur la pertinence de ces résultats expérimentaux pour le cerveau du nouveau-né humain. Il a pourtant été montré que la neurogenèse est un phénomène persistant chez l’homme adulte [45-47]. Tant qu’il n’a pas été établi que la neurogenèse post-hypoxique est un phénomène dommageable, elle porte un espoir de réparation venant à l’appui du concept de l’effet bénéfique d’une stimulation précoce de nourrissons à risque de séquelles d’une hypoxie-ischémie cérébrale. Il est en effet observé qu’un environnement stimulant a des conséquences positives sur les performances neuro-comportementales, et que, chez le rat, les apprentissages ont un effet bénéfique sur la survie neuronale dans certaines structures [48-51].
Les résultats obtenus montrent l’influence du temps d’exposition à l’hypoxie sur le phénomène de neurogenèse qui ne serait plus observable au delà d’un certain délai.
Le caractère transitoire de l’expression de protéines protectrices de la survie cellulaire comme Bcl-2 constitue un argument majeur pour justifier l’urgence d’interventions thérapeutiques appropriées au cours d’épisodes asphyxiques périnatals.
REMERCIEMENTS
Nous remercions Carine Bossenmeyer-Pourié, Gregory Pourié, Valérie Lièvre et Stéphanie Grosjean qui ont mené une grande partie des travaux expérimentaux présentés ici.
BIBLIOGRAPHIE [1] RIVKIN M.J. and VOLPE J.J. — Asphyxia and brain injury. In :
Intensive Care of the Fetus and
Neonate ,. Spitzer A.R. (ed.), Mosby, St Louis, pp. 685-695, 1994.
[2] NYAKAS C., BUWALDA B., and LUITEN, P.G.M. — Hypoxia and brain development.
Prog.
Neurobiol .,1996, 49 , 1-51.
[3] MANERU C., JUNIQUE C., BOTET F., et al. — Neuropsychological long-term sequelae of perinatal asphyxia.
Brain Inj ., 2001, 15 , 1029-1039.
[4] VANNUCCI R. — Experimental biology of cerebral hypoxia-ischemia : relation to perinatal brain damage. Pediatr. Res ., 1990, 27 , 317-326.
[5] HADDAD G.G. and JIANG C. — O2 deprivation in the central nervous system : on mechanisms of neuronal response, differential sensitivity and injury. Prog. Neurobiol .,1993, 40 , 277-318.
[6] MISHRA O.P. — and Delivoria-Papadopoulos M. Cellular mechanisms of hypoxic injury in the developing brain. Brain Res. Bull ., 1999, 48, 233-238.
[7] STTATFORD A. and WHEATHERHALL J.A.C. — The survival of young rats in nitrogen.
J. Physiol ., 1960, 153 , 457-472.
[8] HADDAD G.G. and DONELLY D.F. — O deprivation induces a major depolarization in brains2 tem neurons in the adult but not in the neonate. J. Physiol ., Lond., 1990, 429 , 411-428.
[9] BOWER A.J. — Plasticity in the adult and neonatal central nervous system.
Br. J. Neurosurg ., 1990., 4 , 253-264.
[10] BLACK J.E. — How a child builds its brain : some lessons from animal studies of neural plasticity. Prev. Med ., 1998, 27 , 168-171.
[11] GUBELLINI P., BEN-ARI Y., and GAIARSA J.L. — Activity-and age-dependent GABAergic synaptic plasticity in the developing rat hippocampus. Eur. J. Neurosci ., 2001, 14 , 1937-1946.
[12] KOKAIA Z. and LINDVALL O. — Neurogenesis after ischaemic brain insults. Curr. Opin.
Neurobiol ., 2003, 13, 127-132.
[13] PARENT J.M. — Injury-induced neurogenesis in the adult mammalian brain.
Neuroscientist , 2003, 9 , 261-272.
[14] DAVAL J.L. and VERT P. — Apoptosis and neurogenesis after transient hypoxia in the developing rat brain. Semin. Perinatol. , 2004, 28 , 257-263.
[15] HANSEN M.B., NIELSEN S.E. and BERG., K. — Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Meth ., 1989, 119 , 203-210.
[16] BOSSENMEYER-POURIÉ C., LIEVRE V., GROJEAN S., et al. — Sequential expression patterns of apoptosis- and cell cycle-related proteins in neuronal response to severe or mild transient hypoxia. Neuroscience , 2002, 114 , 869-882.
[17] WOLVETANG E.J., JOHNSON K.L., KRAUER K. et al. — Mitochondrial respiratory chain inhibitors induce apoptosis.
FEBS Letters , 1994, 339 , 40-44.
[18] PARK D.S., MORRIS E.J., GREENE L.A. et al. — G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis.
J. Neurosci., 1997, 17 , 1256-1270.
[19] GROJEAN S., SCHROEDER H., POURIÉ G., et al. — Histopathological alterations and functional brain deficits after transient hypoxia in the newborn rat pup : a long term follow-up.
Neurobiol.
Dis ., 2003, 14 , 265-278.
[20] SHERWOOD N.M. and TIMIRAS P.S. A. — Stereotaxic Atlas of the Developing Rat Brain.
University of California Press, Berkeley, 1970.
[21] ALVAREZ-BUYLLA A., GARCIA-VERDUGO J.M. — Neurogenesis in adult subventricular zone. J.
Neurosci ., 2002, 22 , 629-634.
[22] DAVAL J.L., POURIÉ G., GROJEAN S., et al. — Neonatal hypoxia triggers transient apoptosis followed by neurogenesis in the rat CA1 hippocampus.
Pediatr. Res ., 2004, 55 , 561-567.
[23] BOSSENMEYER C., CHIHAB R., MULLER S., et al. — Hypoxia/reoxygenation induces apoptosis through biphasic induction of protein synthesis in central neurons.
Brain Res ., 1998, 787 ,107- 116.
[24] BOSSENMEYER-POURIÉ C., CHIHAB R., SCHROEDER H., et al. — Transient hypoxia may lead to neuronal proliferation in the developing mammalian brain : from apoptosis to cell cycle completion. Neuroscience, 1999, 91 , 221-231.
[25] SALIBA E. and MARRET S. — Cerebral white matter damage in the preterm infant : pathophysiology and risk factors. Semin. Neonatol., 2001, 6 , 121-133.
[26] CHIHAB R., FERRY C., KOZIEL V., et al. — Sequential activation of activator protein-1 — related transcription factors and JNK protein kinases may contribute to apoptotic death induced by transient hypoxia in developing brain neurons. Mol. Brain Res ., 1998, 63 , 105-120.
[27] BEILHARZ E.J., WILLIAMS C.E., DRAGUNOW M., et al. — Mechanisms of delayed cell death following hypoxic-ischemic injury in the immature rat : evidence for apoptosis during selective neuronal loss. Mol. Brain Res ., 1995, 29 , 1-14.
[28] SIDHU R.S., TUOR U.I., DEL BIGIO M.R. — Nuclear condensation and fragmentation following cerebral hypoxia-ischemia occurs more frequently in immature than older rats. Neurosci Lett ., 1997, 223 , 129-132.
[29] BANASIAK K.J., XIA Y., HADDAD G.G. — Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol ., 2000, 62 , 215-249.
[30] MEIKRANTZ W. and SCHLEGEL R. — Apoptosis and the cell cycle . J. Cell. Biol ., 1995, 58 , 160-174.
[31] TIMSIT S., RIVERA S., OUAGHI P., et al. — Increased cyclin D1 in vulnerable neurons in the hippocampus after ischaemia and epilepsy : a modulator of in vivo programmed cell death ? Eur.
J. Neurosci ., 1999, 11 , 263-278.
[32] HASTINGS N.B., TANAPAT P. and GOULD E. — Neurogenesis in the adult mammalian brain.
Clin. Neurosci. Res ., 2001, 1 , 175-182.
[33] LIE D.C., SONG H., COLAMARINO S.A ., et al. — Neurogenesis in the adult brain : new strategies for central nervous system diseases. Annu.
Rev. Pharmacol. Toxicol .,2004, 44 , 399-421.
[34] MAGAVI S.S., LEAVITT B.R. and MACKLIS J.D. — Induction of neurogenesis in the neocortex of adult mice. Nature. , 2000, 405 , 951-955.
[35] NAKATOMI H., KURIU T., OKABE S., et al. — Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors.
Cell. ,2002, 110, 429-441.
[36] POURIÉ G., BLAISE S., LIÈVRE V., VERT P., DAVAL JL. — Non-lesioning neonatal hypoxia can induce long-term neurogenesis in the rat brain. Pediatr. Res ., 2004, 55 ,25A.
[37] POURIÉ G, BLAISE S., TRABALON M, NÉDÉLEC E., LIÈVRE V., GUÉANT JL, DAVAL JL. — Non lesioning transient hypoxia in the newborn rat induces delayed brain neurogenesis associated with improved memory scores Neurobiology of Disease (soumis).
[38] GOULD E., TANAPAT P., CAMERON H.A. — Adrenal steroïds suppress granule cell death in the devoloping dentate gyrus through and NMDA receptor-dependent mechanism. Brain Res Dev Brain Res ., 1997, 103 , 91-93.
[39] LEMAIRE V., KOEHL M., LE MOAL M. et al. — Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus.
Proc. Nat. Acad. Sci ., 2000, 97 , 1132-1137.
[40] GOULD E., TANAPAT P., MC EVENS B.S., et al. — Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is dimished by stress.
Proc. Nat. Acad. Sci ., 1998, 95 , 3168-3171.
[41] MILLER M.W. — Migration of cortical neurons is altered by gestational exposure to ethanol.
Alcohol Clin. Exp. Res ., 1993, 17 , 304-314.
[42] GIDDAY J.M., FITZGIBBONS J.C., SHAH A.R. et al. — Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat.
Neurosci. Lett ., 1994, 168 , 221-224.
[43] BOSSENMEYER-POURIÉ C., KOZIEL V., DAVAL J.L. — Effects of hypothermia on hypoxia-induced apoptosis in cultured neurons from developing rat forebrain : comparison with preconditioning.
Pediatr. Res ., 2000, 47 , 385-391.
[44] HUTTENLOCHER P.R., RAICHELSON R.M. — Effects of neonatal hemispherectomy on location and number of corticospinal neurons in the rat. Dev. Brain. Res ., 1989, 47 ,59-69.
[45] LIU J., SOLWAY K, MESSING R.O. et al. — Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils.
J. Neurosci ., 1998, 18 , 7768-7778.
[46] DEL BIGIO MR., Proliferative status of cells in adult human dentate gyrus.
Microsc. Res. Tech ., 1999, 45, 353-358.
[47] ERIKSSON PS., PERFILIEVA E., BJORK-ERIKSSON T. et al. — Neurogenesis in the adult human
Hippocampus.
Nat. Med. , 1998, 4 , 1013-1017.
[48] KEMPERMANN G., KUHN H.G. and GAGE F.H. — More hippocampal neurons in adult mice living in an enriched environment. Nature. , 1997, 386 , 493-495.
[49] REMA V. and EBNER F.F. — Effect of enriched environment rearing on impairments in cortical excitability and plasticity after prenatal alcohol exposure. J. Neurosci ., 1999, 19 , 10993-11006.
[50] RISEDAL A., MATTSSON B., DAHLQVIST P., et al. – Environmental influences on functional outcome after a cortical infarct in the rat.
Brain Res. Bull ., 2002, 58 , 315-321.
[51] AMBROGINI P., CUPPINI R., CUPPINI C., et al. — Spatial learning affects immature granule cell survival in adult rat dentate gyrus. Neurosci. Letters., 2000, 286 , 21-24.
DISCUSSION
M. Bernard SALLE
L’ischémie cérébrale est toujours présente dans l’asphyxie prénatale. Avez-vous mesuré le débit sanguin cérébral dans notre modèle in vivo ? La réfrigération a montré (E. Gluckman) que l’apoptose induite par l’hypoxie est diminuée et la neurogénèse augmentée.
Avez-vous une expérience de l’hypothermie dans la prévention des lésions neuronales induites par l’hypoxie ?
Nous n’avons pas pu mesurer le débit sanguin cérébral. Les méthodes avec traceur radio-actif tel que l’iodo-antipyrine marqué supposent un cathétérisme vasculaire impossible chez un rat de sept à dix grammes. En ce qui concerne la prévention des lésions neuronales par l’hypothermie, nous n’avons pas de réponse avec ce modèle. Cependant dans notre laboratoire, nous avons mis en évidence l’effet préventif de l’hypothermie sur des neurones embryonnaires de rat en culture maintenus à 32°. Mais le mécanisme est différent de celui des pré-conditionnements où il y a sur expression de proteines antiapoptotiques comme B-cl2. Cet effet a été reproduit in vivo . L’hypothermie semble agir par réduction du métabolisme énergétique cellulaire.
M. Jacques CAEN
Il s’agit d’abord d’un phénomène répondant à une loi de pathologie générale selon laquelle un préconditionnement avec un stimulus de durée et d’intensité modérées, protège à l’égard du même stimulus de plus forte intensité. Comment peut-on distinguer l’orientation, soit vers une sorte de mort physiologique, l’apoptose, ou vers la nécrose (mort pathologique) ?
Quel est le comportement et le rôle des cellules gliales ? Quelles sont les orientations possibles préventives et thérapeutiques de cette hypoxie neuronale ? Hypothermie (dont les bons résultats semblent avoir été confirmés chez le nouveau-né par Shan Karan (2005) — Apport de glucose + insuline maintenant une normoglycémie — Dotrecogine Cochneider, Nicorandil agissant sur les canaux potassiques ATP sensibles ?
On suppose que l’intensité de l’agression est en cause dans la différence d’orientation vers l’apoptose ou la nécrose, dans une sorte de continuum puisque dans notre modèle in vitro on constate la juxtaposition des deux phénomènes. Cette distinction est faite par l’étude de la chromatine après incorporation du DAPI. Il est par ailleurs, montré que l’excito-toxicité induit préférentiellement des phénomènes de nécrose. Nous n’avons pas de réponse en ce qui concerne les cellules gliales mais elles sont connues pour être plus résistantes à l’hypoxie et jouer un rôle protecteur des neurones. Des travaux montrent aussi qu’après dédifférenciation, elles se comportent comme des cellules souches. Pour ce qui est des orientations thérapeutiques, l’effet bénéfique de l’hypothermie a été montré chez le rat nouveau-né (S. Grojean). Quant à l’apport de glucose ce fut l’objet de grands débats quand, dans les années 1980, R. Myers avait suggéré la possibilité d’une aggravation de l’acidose cellulaire posthypoxique du fait de l’anaérobiose Ceci a été controuvé par Vennucci, et au contraire on a incriminé le déficit en glucose dans la physiopathologie de leucomalacie périventriculaire chez les enfants prématurés (Volpe).
M. Christian NEZELOF
Pourriez-vous commenter le rôle toxique de l’oxygène sur les cellules nerveuses et sur les dangers d’une ‘‘ hyperoxygénation ’’ dans la période néonatale chez les nouveaux-nés prématurés ?
L’oxygène est toxique pour toute cellule vivante et le sujet immature ne dispose pas d’un système anti-oxydant suffisant (super-oxyde —dismutase, catalase, glutathion peroxydase…). Ceci a conduit à recommander l’abstention de l’oxygénation aveugle en réanimation néonatale. Parmi les exemples de cytotoxicité, on peut citer la rétinopathie des
prématurés et la dysplasie broncho-pulmonaire. Sur des neurones en culture, une ré-oxygénation en normoxie avait les mêmes effets qu’une hyperoxie ce qui peut faire considérer qu’une PO 2 à 140 mmHg est déjà excessive pour ces cellules.
M. Georges DAVID
Dans votre réponse à Bernard Salle, vous avez précisé que la réfrigération constitue une protection contre la souffrance hypoxique. Ceci m’évoque les travaux bien anciens d’Henri Laborit qui avaient mis en évidence que cette protection par le froid était potentialisée par les neuroleptiques. Avez-vous testé ces effets des neuroleptiques ?
Nous n’avons pas testé les neuroleptiques mais il est bien démontré que certains d’entre eux agissent par réduction du métabolisme énergétique.
M. Iradj GANDJBAKHCH
Au cours de la chirurgie aortique, il est possible d’arrêter la vascularisation cérébrale jusqu’à 40 minutes à 20 degrés de température. Pensez-vous que la neurogénèse est mise en route dans ces circonstances ? Sinon, pensez-vous que la neurogénèse nécessite la normothermie ?
Nous n’avons pas connaissance d’étude sur le rôle de la température dans la neurogénèse.
Chez la tortue cependant, le réchauffement de l’environnement augmente la prolifération des cellules du système nerveux.
M. Claude DESNUELLE
Chez l’adulte, les modèles animaux d’ischémie/anoxie cérébrale nous apprennent que de nombreux mécanismes sont mis en jeux dès la phase initiale, c’est-à-dire dans les six premières heures qui suivent un accident vasculaire cérébral : déclenchement du programme apoptotique, défaillance énergétique, relargage massif de neurotransmetteurs excitateurs, réaction inflammatoire avec production de radicaux libres, thrombolyse endogène, reperfusion…De cette connaissance, ressort le principe de l’existence d’une fenêtre thérapeutique pour les traitements thrombolytiques ainsi que les principes de traitements neuroprotecteurs pharmacologiques : antagoniste glutamatergique, antagoniste calcique, antiradicaux libres, etc. Dans ce contexte, que sait-on sur le modèle d’hypoxie transitoire pré- senté sur le neurone embryonnaire quant aux mécanismes autres que ceux concernant l’apoptose ?
Nous n’avons pas de réponse pour les neurones en culture mais in vivo , chez le rat, les phénomènes d’excito-toxicité induits par l’hypoxie n’apparaissent qu’après un certain stade de maturation. L’effet des antagonistes comme le MK 801 ou NBQX ne peut se manifester sur des neurones immatures.
M. Francis WATTEL
Que pensez-vous du modèle quasi expérimental de la pendaison manquée (‘‘ dépendue à temps ’’ sans arrêt cardiaque associé au moment de la dépendaison) qui réalise, chez l’homme, un modèle d’hypoxie-ischémie de courte durée et chez lequel l’application de l’oxygénothérapie hyperbare (Dz pur 2.2 ATA. 90 minutes dans un délai inférieur à six heures) amène la disparition de l’ensemble des manifestations cliniques dont la perte de la conscience dans la quasi-totalité des cas (expérience fondée sur une série de trois cents cas observés sur une période de vingt ans) ?
La pendaison peut, en effet, être rapprochée d’un modèle d’ischémie par clampage des carotides chez les animaux par ailleurs exposés à une hypoxie. Nous n’avons pas de réponse sur l’oxygène hyperbare.
Bull. Acad. Natle Méd., 2006, 190, no 2, 469-484, séance du 28 février 2006