
Résumé
Les dérivés antagonistes de la progestérone appartiennent à la grande famille des SPRMs ou ligands spécifiques des récepteurs de la progestérone. Par leur effet très remarquable sur l’endomètre, ils constituent un des grands progrès de la gynécologie puisqu’ils peuvent constituer un traitement remarquable, non chirurgical, des fibromes, en réduisant leur volume d’une part, et en inhibant les saignements qui sont une complication fréquente et invalidante de ces tumeurs bénignes. D’autre part, ces molécules pourraient avoir un intérêt dans le traitement de l’endométriose. Enfin, ces composés peuvent constituer un grand progrès en contraception, puisqu’ils peuvent être utilisés sans estrogène et sans modifier l’environnement estrogènique endogène de la femme.
Summary
Progesterone antagonists belong to the family of selective progesterone receptor modulators. SPRMs already have several applications in women’s health. Their main value lies in their effect on endometrium. For example, they can be used to reduce tumor volume and uterine bleeding before uterine myoma surgery. They are also being evaluated for the treatment of endometriosis, and for estrogen-free contraception. La progestérone, sécrétée durant la phase lutéale par le corps jaune ovarien sous l’effet de la LH, est un régulateur fondamental de la fonction reproductive chez la femme [1, 2] .
Les applications actuelles de la progestérone ou de ses dérivés progestatifs plus puissants sont essentiellement la contraception, et le traitement de la ménopause.
Les stéroïdes qui se lient aux récepteurs de la progestérone appartiennent, en fait, à une classe, plus large, qu’on appelle les modulateurs sélectifs des récepteurs de la progestérone ou SPRMs (Selective Progesterone Receptor Modulators). Les SPRMs peuvent reproduire l’effet de la progestérone, l’antagoniser, voire avoir des effets propres de manière différente selon les organes. Ceci est une particularité remarquable de cette classe thérapeutique.
Mécanisme d’action de la Progestérone et des SPRMs : modulation intracellulaire tissu spécifique de la transcription
La progestérone, ainsi que les différents SPRMs, agissent en se fixant sur des récepteurs nucléaires spécifiques. Le récepteur de la progestérone a été décrit pour la première fois dans l’utérus en 1970 [3]. Le récepteur de la progestérone partage avec les autres récepteurs nucléaires une structure comportant trois domaines fonctionnels principaux. En l’absence de progestérone, le récepteur est « inactif », associé à un ensemble de protéines, notamment les protéines de choc thermique (hsp . En s) présence de progestérone il se produit une série d’événements intracellulaires qui permettent au récepteur lié à la progestérone de moduler la transcription des gènes cibles de celle-ci.
Il existe trois isoformes principales du récepteur de la progestérone issues du même gène : PRA, PRB et PRC (Figure 1), dont la localisation tissulaire et les fonctions ne sont pas identiques. Ces trois isoformes sont codées par le même gène mais la transcription du gène peut être activée en trois sites distincts, aboutissant à la synthèse de trois ARN messagers différents. PR-A ne diffère de PR-B que par le fait qu’il ne contient pas les 164 premiers acides aminés du domaine N terminal. PR-B est activateur de la transcription, PR-A a une activité agoniste moindre in vitro et peut réprimer non seulement l’activité transcriptionnelle du récepteur B mais également celle des autres récepteurs stéroidiens [4]. PRC est exprimée dans le cytoplasme des cellules chorio-amniotiques et placentaires. Cette isoforme, très courte ne peut pas lier l’ADN, elle est donc incapable de moduler directement la transcription des gènes cibles de la progestérone. Elle semble par contre pouvoir lier PRA ou PRB et empêcher leur action biologique. Ce mécanisme serait notamment impliqué dans le déclenchement de la parturition [5].
Le rôle spécifique des isoformes A et B de PR a pu être étudié chez les souris invalidées pour ce récepteur. Chez la souris, le rôle essentiel de PR-A semble être de permettre l’action antiproliférative de la progestérone sur l’endomètre, et celui de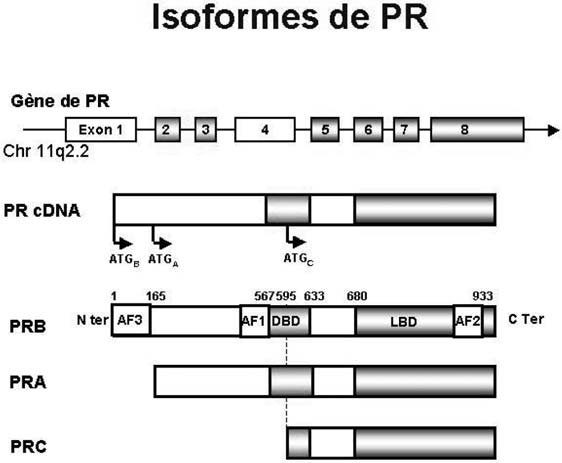
Fig. 1. — Structure primaire des isoformes de PR : LBD domaine de liaison au ligand, DBD domaine de liaison à l’ADN, AF 1, 2, et 3 : domaines d’activation de la transcription 1, 2 et 3, ID :
domaine inhibiteur, ATG A et B : sites d’initiation de la transcription des isoformes A et B.
PR-B de permettre la prolifération normale et la différenciation de l’épithélium mammaire en réponse à la progestérone [6, 7].
Il apparaît donc un premier niveau possible de modulation de l’effet de la progesté- rone dans les tissus cibles, en fonction du rapport des deux isoformes de PR, de manière tissu-sélective. Cependant il existe d’autres niveaux de complexité [6, 8]. La structure de PR est influencée par la nature du ligand avec lequel il interagit. Les agonistes, les antagonistes et les agonistes partiels provoquent chacun des modifications de conformation distinctes dans la structure du récepteur. L’activité biologique du ligand peut être partiellement prédite sur la base de la conformation qu’il provoque sur PR [9]. Par ailleurs, des cofacteurs associés aux récepteurs modulent l’activité transcriptionnelle des récepteurs nucléaires : les coactivateurs augmentent l’activité transcriptionnelle de PR et les corépresseurs la diminuent [10, 11] (Figure 2). Enfin, il a été démontré que PR peut interagir avec d’autres voies de signalisation (jak-stat, fos-jun…), ce qui participe également à la régulation de la transcription des gènes cibles. Ces interactions sont cruciales à connaître pour interpréter les effets rencontrés en clinique.
En dehors de ses effets génomiques, via son récepteur nucléaire, il a été décrit des effets non génomiques de la progestérone, plus rapides [12]. Deux familles de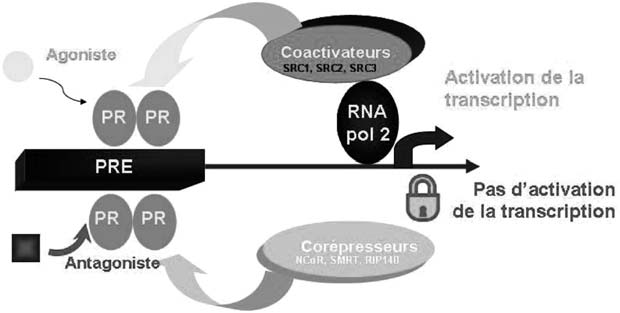
Fig. 2. — Mécanisme moléculaire d’action des ligands du récepteur à la progestérone. En présence d’un ligand agoniste, PR interagit avec des coactivateurs et la transcription est activée. En présence de ligand antagoniste, PR interagit avec des corépresseurs et il n’y a plus d’activation de la transcription.
récepteurs membranaires à la progestérone ont été décrites : la famille, pGMRC1 et 2 et la famille mPRα, mPRβ, mPRγ qui comportent des récepteurs dont la structure s’apparente à celles des récepteurs couplés aux protéines G.
La progestérone peut également se lier sur d’autres récepteurs non spécifiques, comme le récepteur de l’ocytocine, ou le récepteur GABA dans le système nerveux central [13].
Les SPRMs : spectre d’activité et molécules actuelles (Figure 3)
Le premier antagoniste des récepteurs de la progestérone, la mifépristone (initialement RU 486), a été mis au point au début des années 1980 [14]. Elle se comporte comme un antiprogestérone en présence de la progestérone, et a des effets agonistes (progestatifs) en l’absence de progestérone dans certains modèles [15].
Une nouvelle classe de molécules qui fonctionnent comme des modulateurs sélectifs des récepteurs de la progestérone et peuvent avoir une action agoniste ou antagoniste s’est donc développée. L’activité des composés dépend de leur structure et de différents paramètres modulant leur activité [16] : type de tissu, dose, durée du traitement et enfin présence ou absence de progestérone.
La plupart des composés en cours de développement sont des dérivés stéroïdiens.
Différents composés sont actuellement en phase de développement clinique (tableau 1) : la mifépristone est déjà commercialisée, le VA 2914 et l’asoprisnil sont en phase de développement. Enfin, le Proellex® est un composé dont les seules publications sont retrouvées sur le site du fabriquant et dont la structure détaillée et
Fig. 3. — Structure chimique de ligands du récepteur de la progestérone Tableau 1. — Applications cliniques potentielles des SPRMs Administration ponctuelle
Interruption médicale de grossesse*
Induction du travail*
Mort fœtale*
Contraception d’urgence Potentiellement Fécondation in vitro
Administration prolongée
Fibromes, notamment en utilisation préopératoire Métrorragies sous progestatifs Endométriose Contraception Traitement des syndromes dépressifs Cancer du sein (prévention) le mécanisme d’action précis ne sont pas indiquées. Sa structure globale le rapproche du VA2914 Des composés non stéroïdiens sont également à l’étude, à un stade de développement moins avancé.
Utilisation clinique des SPRMs
Les indications potentielles des SPRMs sont très nombreuses et certaines applications sont déjà légales dans de nombreux pays, comme l’interruption de grossesse (volontaire ou thérapeutique), ou proche de la mise sur le marché comme la contraception d’urgence.
Interruption de grossesse
L’interruption de grossesse par méthode médicamenteuse consiste en l’administration de mifépristone et de prostaglandines pour faciliter l’expulsion du conceptus.
La dose de mifépristone préconisée est de 600 mg, mais les études récentes suggèrent que des doses plus faibles [17, 18] sont également efficaces. La mifépristone est également utilisée pour l’interruption thérapeutique de grossesse, la prise en charge des morts fœtales in utero, et le déclenchement du travail [19]. Elle induit la maturation cervicale.
La mifépristone est une molécule bien tolérée. Les seuls effets secondaires décrits sont des douleurs abdominales liées aux contractions utérines, des nausées et des vomissements également induits par les prostaglandines. Des cas d’endométrites associées à des chocs septiques mortels à Clostridium Sordellii ont été décrits [20].
Le lien entre mifépristone et infection n’a pas été démontré mais impose tout de même aux praticiens d’être vigilants dans la surveillance des femmes après interruption médicamenteuse de grossesse.
Contraception
Bien que les contraceptifs oraux estroprogestatifs soient très efficaces, le risque métabolique et thromboembolique associé à l’éthinyl-estradiol, rend nécessaire le développement de méthodes alternatives sans estrogènes. Actuellement les deux solutions disponibles sont les pilules micro progestatives, qui ont l’AMM en contraception, et l’utilisation de progestatifs macro dosés (à dose anti gonadotrope) dans certains contextes particuliers, qui n’ont pas l’AMM en contraception. Le principal effet secondaire des pilules progestatives microdosées est le contrôle insuffisant du cycle avec la survenue de saignements intempestifs. Avec les molécules les plus dosées une inhibition de la sécrétion ovarienne d’estrogènes, et donc une ménopause thérapeutique, inacceptable, survient. Enfin, l’effet potentiellement néfaste des progestatifs sur le sein a été suggéré dans l’étude WHI [21] et, récemment par Fabre [22].
Dans ce contexte, il pourrait être intéressant d’utiliser des molécules ayant un effet antagoniste sur le sein.
Les effets contraceptifs potentiels des SPRMs sont multiples. La mifépristone inhibe la capacité des estrogènes à provoquer le pic de LH et modifie la maturation folliculaire [23]. Elle a également un rôle sur l’endomètre en empêchant l’implantation. Administrée à faible dose en continu la mifépristone bloque l’ovulation, sans empêcher le développement folliculaire [24]. Ceci lui confère un potentiel contraceptif sans induire de déficit estrogénique. De plus un effet direct intra ovarien de la mifépristone a été suggéré [25]. Enfin la mifépristone peut induire une lutéolyse partielle ou totale en fonction de la date d’administration. En phase lutéale précoce, la mifépristone n’entraîne en général qu’une lutéolyse partielle, en phase lutéale tardive, la lutéolyse induite par la mifépristone est en règle générale définitive pour le cycle concerné [26].
Les études cliniques confirment cette inhibition de l’ovulation pour des doses entre 1 et 5 mg/jour [27]. La majorité des patientes présente une aménorrhée pendant le traitement [27].
Plusieurs auteurs ont évalué l’efficacité d’une contraception hebdomadaire par mifépristone. Pei et al [28] n’ont rapporté aucune grossesse sur quatre cent cinquante-six cycles concernant soixante-seize femmes traitées pendant six cycles par 25 ou 50 mg de mifépristone par semaine.
Le VA 2914 a un effet antiovulatoire sans répression de la sécrétion des gonadotrophines aux doses de 5 et 10 mg /j en continu. La dose de 2,5 mg/j ne permet pas d’inhibition de l’ovulation significative. Comme dans le cas de la mifépristone, il persiste une sécrétion estrogénique dans les valeurs de la phase folliculaire physiologique sous traitement [29].
Les effets de l’asoprisnil sur l’axe gonadotrope à moyen terme n’ont pas été décrits à ce jour chez la femme. Au cours d’un mois de traitement, l’asoprisnil inhibe le pic de LH, mais les taux d’estradiol observés en phase folliculaire ne sont pas modifiés [30].
Les SPRMs ont également une place potentielle en contraception orale dans le cadre d’une association avec les progestatifs, pour diminuer les métrorragies. La prescription de mifépristone chez des femmes qui utilisaient un implant progestatif (Norplant) a montré une tendance à la réduction des saignements vaginaux [31].
Une augmentation de la fréquence de l’ovulation a été retrouvée, mais ne supprimerait pas l’effet contraceptif de l’implant [32]. De même l’association d’un SPRM (org 31710) à une contraception microprogestative orale [33] diminue la fréquence des saignements et améliore le contrôle du cycle.
Contraception d’urgence
La mifépristone est utilisée dans de nombreux pays en contraception d’urgence et elle s’est avérée efficace avec peu d’effets secondaires comme « pilule du lendemain ». La mifépristone est actuellement utilisée à la dose 600 mg dans les soixantedouze heures suivant un rapport sexuel non protégé [34]. Cependant des doses plus faibles de 50 mg voire 10 mg semblent aussi efficaces. La mifépristone a une efficacité au moins équivalente à celle de la technique classique dite de Yuzpe (utilisant un estroprogestatif pour administrer 200 μg d’éthinyl estradiol en deux prises), et une meilleure tolérance [35].
L’administration de VA-2914 en phase lutéale du cycle bloque l’activité de la progestérone sur l’endomètre de manière dose dépendante (10, 50, 100, 200 mg) [36].
Le VA 2914 [37], a une double cible à la fois sur la prévention du pic de LH et sur l’implantation, ce dernier mécanisme n’étant pas partagé par le lévonorgestrel. Une étude clinique récente montre qu’il est au moins aussi efficace que le lévonorgestrel et son développement clinique est en cours (Ella®) [37].
Fibromes
Les traitements de référence des fibromes sont les analogues de la GnRH et les progestatifs, générateurs d’effets indésirables (bouffées de chaleur et perte osseuse, troubles du cycle, respectivement).
La mifépristone à la dose de 25 mg par jour induit une aménorrhée, une anovulation et une diminution du volume des fibromes de plus que 50 %. A la dose de 5 mg/j, elle permet également une réduction des symptômes cliniques et une diminution du volume des fibromes avec des effets indésirables minimaux [38].
L’asoprisnil (J867) a également été étudié dans le traitement des fibromes chez les femmes. Les résultats ont montré une suppression dose-dépendante des saignements utérins et la réduction de la dimension des fibromes [39]. La diminution des saignements pourrait s’expliquer par une diminution de la prolifération glandulaire et stromale [40].
Enfin, le Proellex®, antagoniste pur en cours de développement semble également efficace dans cette indication. Ces molécules pourraient avoir une place clé en préparation à la chirurgie, avec une efficacité similaire aux analogues de la GnRH sans risque d’ostéopénie [41].
Endométriose
Les résultats concernant l’endométriose sont plus préliminaires. Les deux complications principales de l’endométriose sont la douleur et l’infertilité. Les études dont nous disposons tendent à montrer que les SPRMs permettent de diminuer les douleurs. La mifépristone à la dose quotidienne de 50 mg semble efficace dans cette indication (amélioration des symptômes et diminution de l’endométriose de 55 %) sans effets indésirables significatifs [42]. Une dose plus faible de mifépristone (5mg par jour pendant six mois) ne permet que le soulagement partiel des symptômes douloureux sans effets sur les lésions. Ces résultats sont donc à confirmer. Le comportement des lésions d’endométriose est spécifique par rapport à l’endomètre eutopique [43] et les effets des SPRMs au niveau des implants endométriosiques sont encore mal connus.
Cancer du sein
L’étude WHI [44] a suggéré des effets délétères potentiels des progestatifs de synthèse. Il est donc actuellement devenu particulièrement légitime de se demander quel pourrait être l’apport des SPRMs dans la prise en charge de diverses pathologies mammaires, y compris en carcinologie mammaire.
Le traitement par la progestérone des lignées humaines de cancer du sein apporte des résultats contradictoires en fonction des contextes cellulaires et de la fonction cellulaire étudiée. Les données disponibles sur la prolifération des cellules tumorales mammaires MCF-7, T47 D et MDA 231 montrent une diminution de la prolifération sous mifépristone [45]. Dans des cellules MCF7 résistantes au tamoxifène, la mifépristone seule ou en association à celui-ci permet d’induire l’apoptose et d’arrêter la prolifération cellulaire.
Le traitement par antiprogestérones (mifépristone et onapristone) permet la réduction des nodules sous cutanés et des métastases ganglionnaires dans un modèle expérimental de greffes de tumeurs mammaires ductales C7-2-HI [46]. Très récemment le groupe de Lee a développé [47] un modèle murin de cancer du sein génétique.
Ces souris ont une invalidation conditionnelle, limitée à la glande mammaire, du gène BRCA1 et de p53. Après administration de mifépristone, aucune des souris traitées ne développe de tumeur à douze mois, alors que les souris contrôles non traitées développent toutes des tumeurs entre quatre et sept mois. Cette donnée est très intéressante car chez la femme les cancers observés en cas de mutation BRCA1 sont moins efficacement prévenus par le tamoxifène que chez les femmes mutées pour BRCA2. La potentialité d’une stratégie alternative d’hormonoprévention est donc du plus grand intérêt.
Des résultats positifs ont été obtenus avec la mifépristone dans le traitement du cancer du sein, chez les patientes devenues résistantes aux thérapies hormonales « conventionnelles »[45]. Cette molécule constitue donc une piste intéressante Enfin, chez les patientes porteuses de mutations des gènes BRCA1 et BRCA2, la possibilité de développer une chimio-prévention est cruciale puisque les seules alternatives disponibles actuellement sont l’annexectomie et la mastectomie prophylactiques. Les données récentes sur la souris pourraient ouvrir de nouvelles perspectives.
Tolérance endométriale au long cours des SPRMs
Les SPRMs sont donc des molécules permettant de maintenir une estradiolémie physiologique tout en ayant une action progestative ou antiprogestative selon les organes. L’existence d’un risque d’effet estrogénique non contrôlé sur l’endomètre doit être précisé. En effet, des aspects d’épaississement endométrial ou d’hyperplasie ont été décrits [48]. Les études histologiques ainsi que les marqueurs utilisés pour juger du statut prolifératif de l’endomètre sont en cours d’évaluation et nécessitent l’établissement par des experts d’un consensus. Il semble en effet que l’aspect observé soit en fait un aspect très spécifique, distinct de l’hyperplasie, observé avec tous les composés de cette classe. Cet aspect endométrial sous SPRMs ne serait ni prolifératif, ni sécrétoire, et serait décrit comme une « dilatation kystique glandulaire » comportant des glandes bordées d’un épithélium inactif. Des aspects « sécré- toires non physiologiques » associant un stroma compact et des glandes tortueuses mais dont l’épithélium n’est pas polarisé ni sécrétoire sont aussi décrits [49].
Le mécanisme précis des effets des SPRMs dans l’endomètre est encore très incomplètement connu et semble faire intervenir notamment des modifications microvasculaires. Une action pro apoptotique ainsi qu’une interaction avec le système micro vasculaire et l’angiogenèse est évoquée [50].
Il est indispensable d’élucider ces effets avant de permettre une utilisation au long cours de ces molécules. Ainsi, pour le moment, le développement de cette classe thérapeutique est axée sur la contraception d’urgence, et le traitement pré opératoire des fibromes.
CONCLUSION
En dépit du désintérêt initial des compagnies pharmaceutiques majeures pour ces composés en raison de leur relation avec la mifépristone (RU 486) et de son image abortive, les études sur les SPRMs se poursuivent.
La question fondamentale à résoudre concerne la tolérance endométriale de ces composés. L’équilibre entre les effets prolifératifs et antiprolifératifs sur l’endomètre sont bien entendu une clé dans l’utilisation à long terme des SPRMs. Il est cependant déjà certain que même si un effet à long terme de type prolifératif sur la muqueuse utérine était observé, il est très vraisemblable que l’administration de progestatifs chaque trois mois pendant une dizaine de jours suffirait à atrophier la muqueuse endométriale. Alternativement, une fenêtre de traitement chaque trois mois aboutirait très probablement au même résultat.
Ces effets semblent pouvoir dépendre de la dose administrée. Compte tenu de l’effet protecteur de la progestérone médié par PRA décrits chez la souris dans l’endomètre il est possible qu’à l’avenir des ligands spécifiques de cette isoforme puissent être développés.
Différentes indications des SPRMs sont envisageables dans un avenir proche : le traitement des fibromes et des saignements associés, la contraception d’urgence.
Plus tard et une fois le problème de la tolérance endométriale correctement étudié, la contraception sans estrogènes pour les femmes à risque pourra être envisagée. A plus long terme des applications au traitement de l’endométriose, des tumeurs hormono — dépendantes pourraient voir le jour.
BIBLIOGRAPHIE [1] Chabbert-Buffet N., Bouchard P. — The normal human menstrual cycle.
Rev. Endocr.
Metab. Disord., 2002, 3 , 173-183.
[2] Ismail P.M., Amato P., Soyal S.M., Demayo F.J., Conneely O.M., O’Malley B.W. et al —
Progesterone involvement in breast development and tumorigenesis-as revealed by progesterone receptor ‘‘ knockout ’’ and ‘‘ knockin ’’ mouse models. Steroids. 2003, 68 , 779-787.
[3] Milgrom E., Atger M., Baulieu E.E. — Progesterone in uterus and plasma. IV — Progesterone receptor(s) in guinea pig uterus cytosol. Steroids, 1970, 16 , 741-754.
[4] Leonhardt S.A., Edwards D.P. — Mechanism of action of progesterone antagonists.
Exp.
Biol. Med., (Maywood) . 2002, 227 , 969-980.
[5] Condon J.C., Hardy D.B., Kovaric K., Mendelson C.R. — Up-Regulation of the Progesterone Receptor (PR)-C Isoform in Laboring Myometrium by Activation of Nuclear Factor{kappa}B May Contribute to the Onset of Labor through Inhibition of PR Function. Mol.
Endocrinol., 2006, 20 , 764-775.
[6] Conneely O.M., Mulac-Jericevic B., Lydon J.P., De Mayo F.J. — Reproductive functions of the progesterone receptor isoforms : lessons from knock-out mice. Mol. Cell Endocrinol., 2001, 179 , 97-103.
[7] Shyamala G., Yang X., Silberstein G., Barcellos-Hoff M.H., Dale E. — Transgenic mice carrying an imbalance in the native ratio of A to B forms of progesterone receptor exhibit developmental abnormalities in mammary glands. Proc. Natl. Acad. Sci., U S A , 1998 , 9 , 696-701.
[8] Giangrande P.H., Kimbrel E.A., Edwards D.P., McDonnell D.P. — The opposing transcriptional activities of the two isoforms of the human progesterone receptor are due to differential cofactor binding. Mol. Cell Bio l., 2000 , 20 , 3102-3115.
[9] Wagner B.L., Pollio G., Leonhardt S., Wani M.C., Lee D.Y., Imhof M.O. et al — 16 alpha-substituted analogs of the antiprogestin RU486 induce a unique conformation in the human progesterone receptor resulting in mixed agonist activity. Proc. Natl. Acad. Sci., U S A, 1996, 93 , 8739-8744.
[10] Lonard D.M., O’Malley B.W. — Nuclear receptor coregulators : judges, juries, and executioners of cellular regulation. Mol. Cel., 2007, 27 , 691-700.
[11] Rowan B.G., O’Malley B.W. — Progesterone receptor coactivators.
Steroids, 2000 , 65 , 545-549.
[12] Falkenstein E., Norman A.W., Wehling M. — Mannheim classification of nongenomically initiated (rapid) steroid action(s). J. Clin. Endocrinol. Metab., 2000, 85 , 2072-2075.
[13] Chabbert-Buffet N.S., D.C., Caraty A., Bouchard P. — Neuroendocrine effects of progestérone. Steroids, 2000, 65 , 613-620.
[14] Philibert D., Deraedt R., Teutsch G. — RU 38486 a potent antiglucocorticoid in vivo . The
VII International Congress of Pharmacology. Tokyo, Japan, 1981.
[15] Gravanis A., Schaison G., George M., De Brux J., Satyaswaroop P.G., Baulieu E.E. — Endometrial and pituitary responses to the steroidal antiprogestin RU 486 in postmenopausal women. J. Clin. Endocrinol. Metab., 1985, 60, 156-163.
[16] Spitz I.M. — Progesterone antagonists and progesterone receptor modulators.
Expert Opin.
Investig. Drugs, 2003, 12, 1693-1707.
[17] Who Task Force on Post — Ovulatory Methods of Fertility Regulation : Termination of Early Human Pregnancy with RU 486 (Mifepristone) and the prostaglandin analogue sulprostone : a multi — center, randomized comparison between two treatment regimens.
Hum. Reprod., 1989, 4 , 718- 725.
[18] Christin-Maitre S., Bouchard P., Spitz I.M. — Medical Termination of Pregnancy.
New
Engl. J. Med. , 2000, 342 , 946-956.
[19] Chabbert-Buffet N., Meduri G., Bouchard P., Spitz I. — Selective Progesterone Receptor Modulators and Progesterone Antagonists : mechanism of action and clinical applications.
Human Reprod. Updates, 2005 , 11 , 293-307.
[20] Couzin J. — Infectious disease. RU-486-linked deaths open debate about risky bacteria.
Science , 2006, 312 , 986.
[21] Effects of conjugated equine estrogen in postmenopausal women with hysterectomy. The Women’s Health Initiative randomized controlled trial. JAMA , 2004, 291 , 1701-1712.
[22] Fabre A., Fournier A., Mesrine S., Desreux J., Gompel A., Boutron-Ruault M.C. — Oral progestagens before menopause and breast cancer risk. Br. J. Cancer, 2007, 96 , 841-844.
[23] Luukkainen T., Heikinheimo O., Haukkamaa M., Lahteenmaki P. — Inhibition of folliculogenesis and ovulation by the antiprogesterone RU 486. Fertil. Steril. ,1988, 49 , 961-963.
[24] Baird D.T., Brown A., Critchley H.O., Williams A.R., Lin S., Chen G. — Effect of long-term treatment with low-dose mifepristone on the endometrium. Hum. Reprod., 2003 , 18, 61-68.
[25] Qiu X., Sun X., Christow A., Stabi B., Gemzell-Danielsson K. — Action of mifepristone on the expression of insulin-like growth factor binding protein-1 mRNA and protein during the early luteal phase in the human oviduct. Fertil. Steri., 2003, 80 Suppl 2, 776-782.
[26] Dubois C., Ulmann A., Baulieu E.E. — Contragestion with late luteal administration of RU 486 (Mifepristone). Fertil. Steril., 1988, 50 , 593-596.
[27] Brown A., Cheng L., Lin S., Baird D.T. — Daily low-dose mifepristone has contraceptive potential by suppressing ovulation and menstruation : a double-blind randomized control trial of 2 and 5 mg per day for 120 days. J. Clin. Endocrinol. Metab. , 2002, 87 , 63-70.
[28] Pei K., Xiao B., Jing X., Lu S., Wei L., Zhao H. — Weekly contraception with mifepristone.
Contraception , 2007, 75 , 40-44.
[29] Chabbert-Buffet N., Pintiaux-Kairis A., Bouchard P. — Effects of the progesterone receptor modulator VA2914 in a continuous low dose on the hypothalamic-pituitary-ovarian axis and endometrium in normal women : a prospective, randomized, placebo-controlled study.
Journal of Clinical endocrinology and metabolism , 2007 , 92(9) , 3582-3589.
[30] Chwalisz K., Elger W., Stickler T., Mattia-Goldberg C., Larsen — The effects of 1-month administration of asoprisnil (J867), a selective progesterone receptor modulator, in healthy premenopausal women. Hum. Reprod. , 2005, 2, 1090-1099.
[31] Glasier A.F., Wang H., Davie J.E., Kelly R.W., Critchley H.O. — Administration of an antiprogesterone up-regulates estrogen receptors in the endometrium of women using Norplant : a pilot study. Fertil. Steril. , 2002, 77, 366-372.
[32] Cheng L., Zhu H., Wang A., Ren F., Chen J., Glasier A. — Once a month administration of mifepristone improves bleeding patterns in women using subdermal contraceptive implants releasing levonorgestrel. Hum. Reprod. , 2000, 15, 1969-1972.
[33] Gemzell-Danielsson K., Van Heusden A.M., Killick S.R., Croxatto H.B., Bouchard P., Cameron S. et al . — Improving cycle control in progestogen-only contraceptive pill users by intermittent treatment with a new anti-progestogen.
Hum. Reprod., 2002, 17 , 2588-2593.
[34] Psychoyos A., Nikas G., Sarantis L., Gravanis A. — Hormonal anti-implantation agents :
antiprogestins. Hum. Reprod., 1995 , 10 Suppl 2, 140-150.
[35] Randomised controlled trial of levonorgestrel versus the Yuzpe regimen of combined oral contraceptives for emergency contraception. Task Force on Postovulatory Methods of Fertility Regulation . Lancet ,1998, 352 , 428-433.
[36] Passaro M.D., Piquion J., Mullen N., Sutherland D., Zhai S., Figg W.D. et al . — Luteal phase dose-response relationships of the antiprogestin CDB-2914 in normally cycling women.
Hum. Reprod., 2003, 1820-1827.
[37] Creinin M.D., Schlaff W., Archer D.F., Wan L., Frezieres R., Thomas M. et al . —
Progesterone receptor modulator for emergency contraception : a randomized controlled trial.
Obstet. Gyneol. , 2006, 108, 1089-1097.
[38] Eisinger S.H., Meldrum S., Fiscella K., Le Roux H.D., Guzick D. — Low-dose mifepristone for uterine leiomyomata. Obstet. Gynecol., 2003, 101 , 243-250.
[39] Chwalisz K., Larsen L., Mattia-Goldberg C., Edmonds A., Elger W., Winkel C. — A randomized, controlled trial of asoprisnil, a novel selective progesterone receptor modulator, in women with uterine leiomyomata. Fertil. Steril. , 2007 , 87 , 1399-1412.
[40] Williams A.R., Critchley H.O., Osei J., Ingamells S., Cameron I.T., Han C., Chwalis Z. — The effects of the selective progesterone receptor modulator asoprisnil on the morphology of uterine tissues after 3 months treatment in patients with symptomatic uterine leiomyomata.
Hum. Reprod. , 2007, 27, 1696-1704.
[41] Grow D.R., Willims R.F., Hsiu J.G., Hodgen G.D. — Antiprogestin and/or gonadotropinreleasing hormone agonist for endometriosis treatment and bone maintenance : a 1-year primate study. J. Clin. Endocrinol. Metab. , 1996 , 81 ,1933-1939.
[42] Kettel L.M., Murphy A.A., Morales A.J., Ulmann A., Baulieu E.E, Yen S.S. — Treatment of endometriosis with the antiprogesterone mifepristone (RU486). Fertil. Steril. ,1996, 65 , 23-28.
[43] Giudice L.C., Kao L.C. — Endometriosis. Lancet , 2004, 364 , 1789-1799.
[44] Risks and benefits of estrogen plus progestin in healthy postmenopausal women : principal results from the Women’s Health Initiative Randomized controlled trial. JAMA , 2002 , 288 , 321-333.
[45] Klijn J.G., Setyono Han B., Foekens J.A. — Progesterone antagonists and progesterone receptor modulators in the treatment of breast cancer. Steroids, 2000, 65 , 825-830.
[46] Vanzulli S.I., Soldati R., Meiss R., Colombo L., Molinolo A.A., Lanari C. — Estrogen or antiprogestin treatment induces complete regression of pulmonary and axillary metastases in an experimental model of breast cancer progression. Carcinogenesis , 2005 , 26 , 1055-1063.
[47] Poole A.J., Li Y., Kim Y., Lin S.C., Lee W.H., Lee E.Y. — Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science, 2006, 314, 1467- 1470.
[48] Eisinger S.H., Meldrum S., Fiscella K., Le Roux H.D., Guzick D.S. — Low-dose mifepristone for uterine leiomyomata. Obstet. Gynecol., 2003, 101 , 243-250.
[49] Horne F.M., Blithe D. — Meeting Report. Progesterone Receptor Modulators and the Endometrium : Changes and Consequences. Hum. Reprod. Update, 2007, 13(6) , 567-580.
[50] Chen W., Ohara N., Wang J., Xu Q., Liu J., Morikawa A. et al . — A novel selective progesterone receptor modulator asoprisnil (J867) inhibits proliferation and induces apoptosis in cultured human uterine leiomyoma cells in the absence of comparable effects on myometrial cells. J. Clin. Endocrinol. Metab. , 2006, 91 , 1296-1304.
DISCUSSION
M. Jean-Louis CHAUSSAIN
Quel est le mécanisme de l’efficacité dans le cancer du sein dans la mesure où la sécrétion d’oestrogène par l’ovaire est conservée ?
L’effet des antiprogestérones ou Modulateurs des Récepteurs de la Progestérone (PRM) est un effet de type antiprolifératif. Celui-ci a été mis en évidence dans les myomes utérins où leur administration induit une apoptose accélérée. Au niveau du sein, l’étude de Poole et col . montre un effet suppresseur de tumeurs chez des souris génétiquement modifiées pour faire un cancer du sein. Ainsi l’effet antitumoral des PRMs ne nécessite pas la présence d’une sécrétion ovarienne normale.
M. Claude SUREAU
Peut-il exister des indications obstétricales (évacuation utérine, déclenchement du travail) à ces molécules ?
A ce jour tous les PRMs que j’ai présentés (Ulipristal Asoprisnil, Proellex…) se différencient de la mifepristone (RU 486) par leur faible activité abortive. Ils sont, de plus, utilisés à doses faibles, trente à quarante fois plus faibles que la dose légale de mifépristone. De ce fait ils n’ont pas d’applications obstétricales ou abortives.
M. Jean-Luc de GENNES
Après cette excellente communication, et non moins excellente illustration, je désirerais quelques précisions sur les niveaux des oestrogènes produits par les ovaires. Ces niveaux sont-ils ceux de la phase folliculaire ? Et n’y a-t-il pas des inconvénients au maintien permanent de ces niveaux d’oestrogènes ?
Chez une femme traitée par modulateur des récepteur de la progestérone, la sécrétion endogène d’estradiol n’est pas réduite, ce qui explique leur parfaite tolérance, notamment l’absence de diminution de la masse osseuse, constamment observée avec les analogues de la GnRH. En administration continue, comme vous le suggérez, il existe des modifications particulières de l’endomètre dénommées PAECs (PRM Associated Endometrial Changes). Ces modifications ne ressemblent en rien à l’aspect classique d’hyperplasie, état pré cancéreux bien identifié. Les PAECs sont essentiellement constitués d’une dilatation glandulaire kystique, responsable d’une augmentation de l’épaisseur endomé- triale. Ces modifications apparaissent banales, néanmoins l’usage continu sur de longues périodes des PRMs devra attendre une clarification de ces aspects au cours d’études cliniques de longue durée. Dans l’immédiat, les indications qui apparaîtront sur le marché pharmaceutique devront se réduire à, au plus, trois mois de traitement.
M. Edwin MILGROM
Pouvez-vous discuter le problème de l’activité antiglucocorticoïde des anciens antagonistes et des nouveaux SPRM ? Toutes les données théoriques sur les formes A ou B du récepteur, sur l’interaction avec les co-activateurs et les corépresseurs, fonctionnent bien dans le tube à essai. Mais tous les SPRM obtenus jusqu’ici l’ont été de façon empirique : synthèse chimique puis sélection avec des tests pharmacologiques classiques. Qu’en pensez vous ?
Dans l’avenir, les études structurales (diffraction des rayons X par les cristaux de PR contenant différents ligands) pourront-elles aboutir à une synthèse logique de nouveaux SPRM ?
En premier lieu, les PRMs qui sont développés pour la contraception d’urgence, le traitement des saignements utérins, ou pour le traitement des fibromes n’ont pas d’effet antiglucocorticoide connu. Seule la mifepristone possède un effet antiglucocorticoide significatif et est même utilisée dans le traitement du syndrome de Cushing. Toutefois, ce même composé utilisé à faibles doses (2-50 mg par jour) n’a pas d’effets significatifs de ce type. La question sur l’impact des études structurales sur la synthèse de nouveaux PRMs est intéressante. Des études récentes ont abordé ce problème. Comme le suggère la question, les études in vitro sur l’identification et l’étude des corépresseurs et des coactivateurs a apporté une tentative d’explication du mécanisme d’action des PRMs. De manière simpliste, il apparaît que les agonistes recrutent des coactivateurs tandis que les plus antagonistes recrutent des corépresseurs. Toutefois ce mécanisme n’explique pas que les PRMs ont tous les mêmes effets sur l’endomètre nonobstant leur capacité à recruter tel ou tel coactivateur ou corépresseur. Beaucoup reste à comprendre, les études structurales sont à cet égard certainement très utiles. La reconnaissance des acides aminés impliqués dans la liaison des PRMs au sein de l’hélice 12 du domaine de liaison du ligand dans la molécule de PR permettra sûrement d’aider à la synthèse de nouveaux PRMs à effet antagoniste.
* Endocrinologie, Hôpital Saint Antoine, 184 rue du Faubourg Saint Antoine — 75012 Paris. ** EA 1533 UPMC Univ. Paris 06 *** Gynécologie Hôpital Tenon. Tirés à part : Professeur Philippe Bouchard, même adresse Article reçu le 24 décembre 2007, accepté le 3 mars 2008
Bull. Acad. Natle Méd., 2008, 192, no 6, 1159-1173, séance du 10 juin 2008