
Résumé
Les études des enfants atteints de syndrome dysmorphique permettent d’envisager dans certains cas, un diagnostic étiologique et donc un pronostic et une prise en charge adaptés de l’enfant, ainsi qu’un conseil génétique dans la famille. Ces syndromes correspondent à des anomalies du développement embryonnaire d’origine génétique. Les analyses génétiques permettent actuellement l’identification de gènes impliqués dans le développement embryonnaire. La reconnaissance de certaines familles de gènes du développement a progressé grâce à l’identification de patients et aux progrès de la génétique médicale. Ces gènes souvent méconnus jusque-là dans le domaine du développement, sont impliqués dans la morphogenèse normale et anormale chez l’homme. Un grand nombre de ces gènes codent pour des facteurs de transcription, régulant l’expression d’autres gènes au cours du développement embryonnaire.
Summary
Studies of children with developmental abnormalities of genetic origin are necessary for accurate diagnosis, prognostication, patient management, and genetic counseling. Such studies can also help to identify genes involved in normal and abnormal morphogenesis, which often act as patterning genes and are also potential oncogenes. Many encode transcription factors that regulate other genes during embryonic development. Le développement embryonnaire humain commence par un seul œuf fertilisé, ainsi la programmation des différentes phases de division, de différenciation et de croissance cellulaires est particulièrement complexe pour aboutir à un enfant normal. Les gènes impliqués dans le développement peuvent être de structure, de fonction ou de famille très variées, comme des ligands, des récepteurs, des transporteurs d’anions, des molécules d’adhésion cellulaire ou encore impliqués dans la transduction du signal. L’étude de mutants de drosophile avec un défaut de la segmentation ou du développement du corps par Nüsslein-Volhard et coll. [1] a permis de distinguer trois groupes de gènes impliqués dans le développement embryonnaire (gap, pair-rule , polarité segmentaire). L’identification de ces gènes a indiqué que la plupart d’entre eux codaient pour des facteurs de transcription. Ces derniers, très conservés dans l’évolution, régulent l’expression des gènes en se liant par des motifs spécifiques à l’ADN. La théorie d’une base moléculaire du développement repose sur la caracté- risation de chaque type cellulaire par son patron d’expression génomique temporospatiale, c’est-à-dire « quels gènes seront exprimés à quel moment ? ». Le niveau principal de contrôle de l’expression des gènes se situe au niveau de la transcription et les facteurs de transcription régulent le développement embryonnaire par le contrôle de gènes cibles. La reconnaissance clinique de syndromes dysmorphiques et malformatifs chez l’enfant en tant qu’anomalies du développement embryonnaire d’origine génétique a permis d’identifier des mutations dans de nombreux gènes du développement codant pour des facteurs de transcription chez l’homme [2]. Cette synthèse est focalisée sur les anomalies du développement liées à des mutations dans des gènes impliqués dans la régulation de la transcription. Les facteurs de transcription sont des composants de complexes multiprotéiques nécessaires pour l’initiation de l’ARN polymérase II dans la transcription. Les facteurs de transcription sont caractérisés par des séquences d’acides aminés conservés parmi les espèces, des domaines spécifiques de liaison à l’ADN avec des motifs caractéristiques et dans la plupart des cas, une activité fonctionnelle sous forme dimérique. Un facteur de transcription se compose généralement d’un domaine de liaison à l’ADN et d’un domaine d’activation de la transcription. Les facteurs de transcription peuvent être classés sur la base de leurs motifs structuraux qui se lient à l’ADN et dont les plus fréquents sont : doigt de zinc, homeodomaine, glissière de leucine (« leucine zipper »), hélice-tour-hélice, boîtes HMG (high mobility group) et boîtes T (Figs. 1, 2, 3).
LES GÈNES HOX ET LES ANOMALIES DES MEMBRES
Anomalies dues à des mutations des gènes HOX
Les gènes à homéodomaine ont initialement été identifiés par leur rôle majeur dans le développement de la segmentation de la drosophile grâce aux mutations homéo-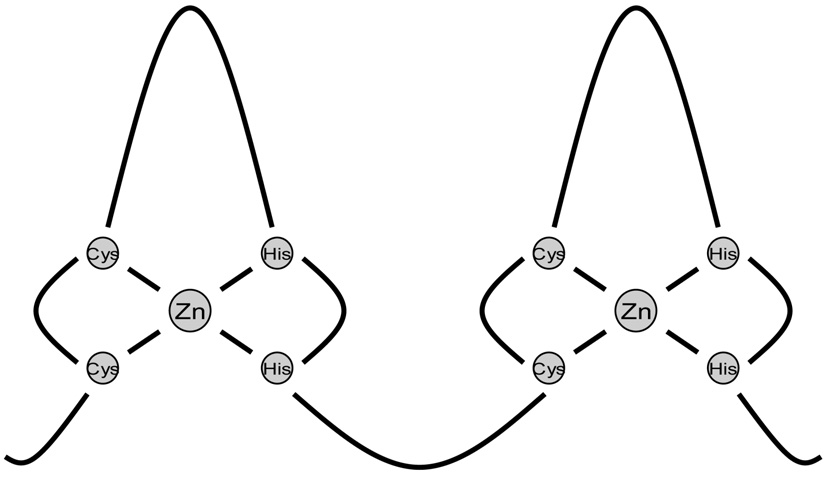
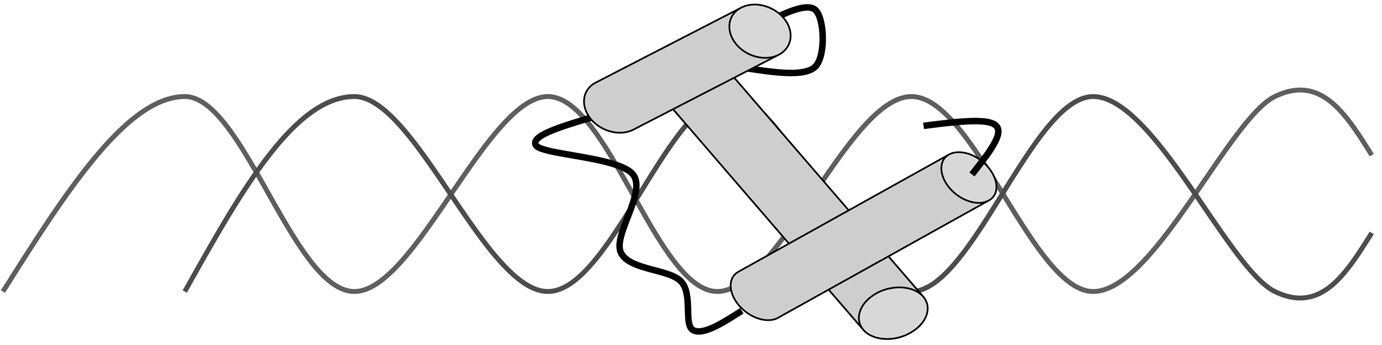
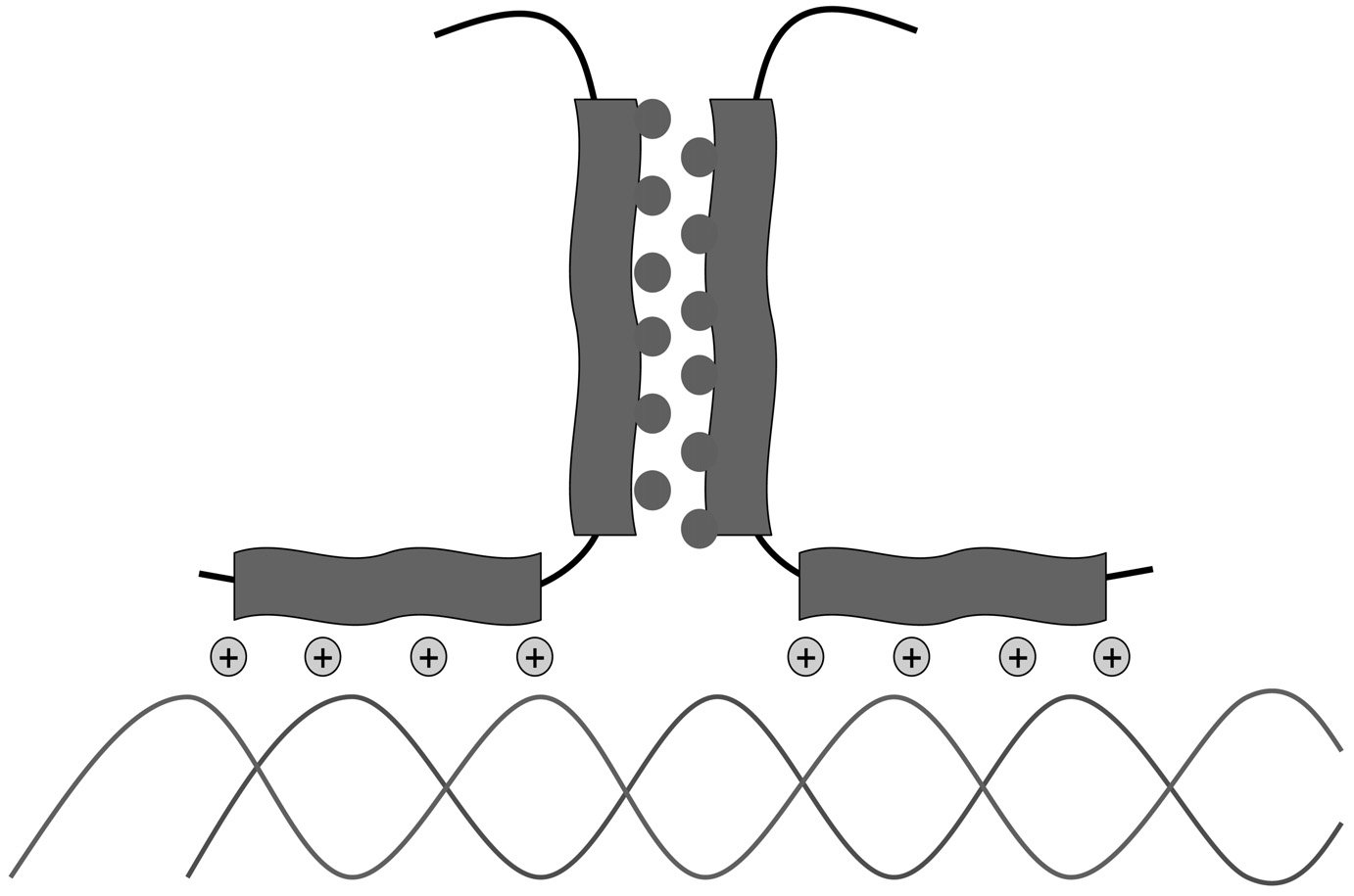
Fig. 1. — Motif de liaison de l’ADN type doigt de zinc Fig. 2. — Motif de liaison à l’ADN type hélice-tour-hélice Fig. 3. — Motif de liaison à l’ADN de type glissière de leucine tiques. Les gènes Hox remarquablement conservés dans l’évolution ont une expression spatio-temporelle colinéaire du développement de l’axe antéro-postérieur de l’embryon, notamment au cours du développement des membres. Des mutations des gènes Hox peuvent entraîner le développement de pattes à la place d’antennes chez la mouche. Ces gènes se caractérisent par un homéodomaine carboxy-terminal qui correspond à un motif de 60 AA se fixant à l’ADN. Chez la drosophile, le complexe Hom-C se répartit en deux groupements, bithorax et antennapedia. Chez les vertébrés, il existe 39 gènes HOX organisés en quatre complexes (HOXA, HOXB, HOXC, HOXD). Des données moléculaires récentes indiquent que les protéines Hox interagissent avec d’autres protéines à homéodomaine et fonctionnent dans des complexes multiprotéiques [3].
Ce rôle clef de la morphogénèse a longtemps fait suspecter le rôle pathogène de mutations des gènes HOX dans des phénotypes malformatifs sévères chez l’homme et chez la souris. Plusieurs études ont démontré que les gènes HOX, particulièrement ceux des complexes A et D, sont impliqués dans des anomalies des extrémités, en regard notamment des malformations importantes des pattes obtenues dans les modèles murins par mutagenèse dirigée.
Le gène HOX D13 a été impliqué dans la synpolydactylie (syndactylie type II) chez l’homme par un mécanisme de gain de fonction avec expansion d’une polyalanine dans la région amino-terminale du gène en dehors de l’homéodomaine [4]. La synpolydactylie est une affection dominante caractérisée chez les hétérozygotes par des syndactylies III/IV des doigts et IV/IV des orteils avec la duplication d’un rayon dans la syndactylie membraneuse. Le phénotype homozygote est plus sévère et associe la transformation de métacarpes et de métatarses en os courts du carpe et du tarse. L’expansion de polyalanine se situe généralement entre sept et quatorze résidus ; des expansions plus grandes semblent produire un phénotype plus sévère.
Des garçons affectés dans une famille avec une grande expansion ont également un hypospadias consistant avec l’expression de HOX D13 dans le tubercule génital [5].
Des expansions de groupement alanine ont également été décrites dans des syndromes humains liés à d’autres facteurs de transcription comme CBFA1 (Core-Binding Factor Alpha Subunit 1) ou ZIC2. Des mutations faux-sens dans l’homéodomaine du gène HOX D13 ont été associées aux brachydactylies de type D et E [6].
Des mutations du gène HOXA13 ont été identifiées dans des familles présentant un syndrome main-pied-utérus, qui associe des anomalies mineures des extrémités (raccourcissement du premier et du cinquième rayon des mains et des pieds), et des malformations génito-urinaires [7].
Anomalies dues à des mutations dans d’autres gènes à homéodomaine
Le premier exemple en pathologie humaine d’une mutation dans un gène à homéodomaine fut l’identification d’une mutation du gène MSX2 (Muscle Segment Homeobox 2) dans une famille où ségrégeait sur un mode dominant une craniosté- nose type Boston [8]. Des mutations de MSX2 ont depuis été identifiées dans des cas de foramen pariétal isolé sans craniosténose, ce qui semble traduire la variabilité d’un spectre clinique lié à une différenciation ostéogénique anormale du crâne. Les gènes MSX font partie d’une petite famille de gènes homologues du gène msh (muscle-segment homeobox) de Drosophile. Une mutation hétérozygote de l’homéodomaine du gène humain MSX1 est responsable d’une agénésie sélective des secondes prémolaires et des troisièmes molaires [9].
Certains patients atteints de schizencéphalie sont porteurs de mutations du gène EMX2 (Empty Spiracles 2, homologue de Drosophile), un gène à homéoboîte impliqué dans le développement du système nerveux central [10]. La schizencéphalie est une malformation rare chez l’homme caractérisée par une fente des hémisphères cérébraux. Les mutations de EMX2 sont préférentiellement identifiées dans les cas de schizencéphalie sévère (type II) avec un large défaut parenchymateux, une fente holohémisphérique et un retard mental.
Anomalies des membres et gènes à boîte T
Le syndrome de Holt-Oram qui associe des malformations cardiaques et des anomalies du rayon radial des membres supérieurs comme des pouces triphalangiens est dû à des mutations dans le gène TBX5 (T-Box 5) [11]. Les anomalies des membres supérieurs rencontrées dans le syndrome de Holt-Oram sont volontiers asymétriques, voire unilatérales, et les anomalies cardiaques peuvent être détectées uniquement sur l’analyse de l’électrocardiogramme. L’expressivité du syndrome est variable, pouvant se limiter à l’atteinte d’un seul des deux appareils et parfois différente chez certains membres atteints de la même famille. Le gène TBX5 est un des membres de la famille Brachyury (T) à boîte T dont on connaît plus de 20 gènes.
Il a été identifié des mutations dans un autre gène de cette famille, TBX3, dans un syndrome rare associant des anomalies des extrémités, de l’appareil génital et des glandes mammaires [12]. Le gène TBX1 est impliqué dans le déterminisme de la microdélétion 22q11.2 et les mutations à type de gain de fonction ou de perte de fonction entraînent le même phénotype que la délétion 22q11 [13]. Des phénotypes ont aussi été associés aux gènes TBX4 (chondrodysplasie à petites rotules) et TBX22 (fente palatine et ankyloglossie).
LES GÈNES PAX
L’identification initiale des facteurs de transcription comme gènes du développement a révélé que la plupart d’entre eux se liaient à l’ADN par le domaine homéo de 61 acides aminés (AA) ou par un domaine de 128 AA (domaine paired). Les gènes contenant des domaines paired ont été dénommés gènes PAX (Paired Box gene ou Paired domain gene HuP2). Le motif paired a une propriété de liaison par deux motifs hélice-tour-hélice et certains gènes PAX (PAX3/PAX7, PAX4/PAX6) ont aussi un deuxième domaine de liaison à l’ADN de type homeoboîte. La famille de gènes PAX a été identifiée par homologie avec des gènes de segmentation de la Drosophile ( paired, gooseberry-proximal et gooseberry-distal ). Chez la souris et l’homme, 9 gènes PAX ont été identifiés et dénommés PAX1 à PAX9. Ils sont très bien conservés au cours de l’évolution et existent également chez le poisson-zèbre et les céphalopodes. Ils ont été classés en 4 groupes qui partagent une organisation commune de motifs protéiques (PAX1/PAX9, PAX2/PAX5/PAX8, PAX3/PAX7, PAX4/PAX6) et un patron d’expression similaire au cours de l’embryogenèse [14].
Les gènes PAX ne sont pas regroupés dans le génome, mais sont localisés sur différents chromosomes.
Cinq gènes PAX sont actuellement connus pour être impliqués dans des dysmorphies chez l’homme (Table 1). Les anomalies du développement sont dues à une haplo-insuffisance alors que des mécanismes de gain de fonction ont été impliqués dans certaines formes de cancers. Par exemple, un gène chimérique (fusion de 2 gènes) impliquant PAX3 a été identifié dans des cas de rhabdomyosarcome chez l’enfant [15]. Nous ne citerons que l’exemple de PAX3.
Table 1. — Maladies humaines dues à des mutations dans les gènes PAX Gène
Localisation
Phénotype délétère chez l’homme chromosomique
PAX2 10q25 Colobomes du nerf optique et anomalies rénales PAX3 2q35 Syndrome de Waardenburg (types I et III) PAX6 11p13 Aniridie Anomalie de Peter’s Cataracte Kératite Dysplasie fovéale PAX8 2q12-q14 Dysgénésie thyroïdienne (hypoplasie, ectopie) PAX9 14q12 Hypodontie Des mutations dans le gène Pax3 sont responsables du mutant Splotch chez la souris qui associe anomalies de la pigmentation (tache blanche sur l’abdomen, la queue ou l’extrémité des pattes) et défauts de fermeture du tube neural. La reconnaissance du phénotype murin a permis d’identifier des mutations dans le gène homologue humain PAX3 chez des patients atteints de syndrome de Waardenburg de type 1 (SW1) [16]. Le SW1 touche principalement les tissus dérivés des crêtes neurales, associant une dystopie des canthi (déplacement exagéré des angles internes des yeux), une pigmentation anormale (hérérochromie irienne, mèche de cheveux blancs, taches achromiques), une surdité de perception par atteinte des cellules de Corti qui ont pour origine les crêtes neurales, et des critères dysmorphiques inconstants (synophris, racine du nez large). Deux mutations homozygotes de PAX3 ont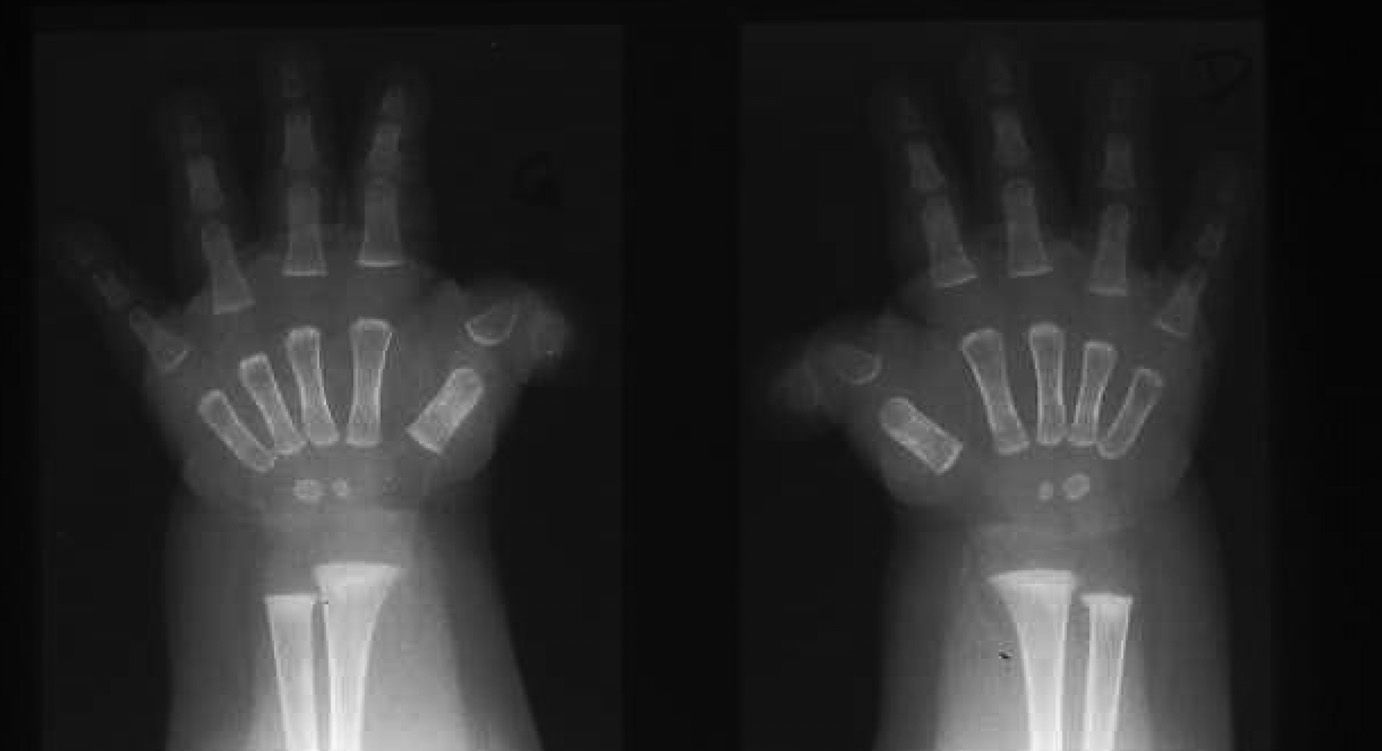
Fig. 4. — Radiographie des mains de face d’un enfant atteint de syndrome de Rubinstein-Taybi montrant l’aspect des pouces larges en déviation angulaire.
été rapportées chez l’homme avec un phénotype très similaire à celui de la souris dans un cas (exencéphalie, arthrogrypose) et dans l’autre, un aspect de syndrome de Waardenburg de type 3 avec une dystopie des canthi sévère, un albinisme partiel et une anomalie sévère des membres supérieurs. Le SW3 (ou syndrome de KleinWaardenburg) associe au phénotype de SW1 des anomalies des membres supérieurs et est maintenant connu comme étant lié à des mutations de PAX3, même dans le cas original décrit par Klein. Une autre mutation de PAX3 (Asn47Lys) a été retrouvée dans une famille avec un syndrome craniofacial avec surdité et anomalies des mains.
Le SW de type 2 se distingue du SW1 par l’absence de dystopie des canthi. Ce signe dysmorphique a permis de classer les patients dans deux groupes cliniques distincts.
Le SW2 est maintenant connu comme étant lié dans certains cas à des mutations du gène MITF (Microphtalmia associated Transcription Factor) sur le chromosome 3q12.3-p14.1 [17], contrairement à SW1. Cette différence mineure de morphologie faciale entre les patients a permis de distinguer des phénotypes corrélés à des génotypes distincts. Le gène MITF code pour un facteur de transcription de type hélice-boucle-hélice et glissière de leucine, exprimé dans la peau adulte et chez l’embryon la rétine, la vésicule optique et le follicule pileux.
SYNDROMES DYSMORPHIQUES AVEC RETARD MENTAL
Exemple du syndrome de Rubinstein-Taybi
Le syndrome de Rubinstein-Taybi (SRT) se caractérise par un retard de développement psychomoteur, un retard de croissance, des pouces et des gros orteils larges avec possible déformation angulaire et duplication de la dernière phalange (Fig.4), et une dysmorphie faciale. L’aspect facial classique chez l’enfant associe une orientation en bas et en dehors des fentes palpébrales, des replis épicanthiques, un ptosis, un strabisme, une voûte palatine très ogivale, des oreilles bas implantées en rotation postérieure, et un nez marqué avec racine protruse, septum long et saillant, et columelle courte. Le phénotype facial est évolutif avec un aspect moins marqué chez le nouveau-né. Cette dysmorphie faciale ne devient caractéristique que dans l’enfance. D’autres anomalies (oculaires, génito-urinaires, osseuses, cutanées, cardiaques, digestives…) ont été rapportées ainsi qu’un risque tumoral accru. Les tumeurs rapportées dans le cadre du SRT sont surtout des tumeurs du système nerveux (médulloblastome, gliome, neuroblastome, méningiome), mais d’autres tumeurs ont également été décrites dont quelques cas de leucémies aiguës lymphoblastiques ou myéloblastiques. Un excès de cicatrisation anormale type chéloide est aussi connu chez les patients atteints de SRT.
La grande majorité des cas sont sporadiques, mais quelques rares familles de transmission dominante ont été décrites. Une microdélétion du chromosome 16p13.3 est détectable par hybridation in situ en fluorescence (FISH) et identifiée seulement chez environ 12 % des patients atteints de SRT [18]. Des mutations ponctuelles non-sens et des délétions, suggérant un mécanisme d’haplo-insuffisance, ont été mises en évidence dans le gène CBP (ou CREB) qui code pour la protéine CREBBP (CREB Binding Protein). Des mutations ont plus récemment été identifiées chez des patients dans le gène EP300. CBP et son homologue EP300 sont des coactivateurs de transcription qui interviennent dans diverses voies de transduction de signal déclenchées en réponse à des stimuli extracellulaires, et jouent un rôle important dans la régulation de la croissance et de la différenciation cellulaire [19].
CBP est notamment un coactivateur de l’expression de gènes possédant des élé- ments cis-régulateurs CRE (cAMP response elements, ou cAMP-regulated enhancer) régulés par l’AMP cyclique. La CBP se lie en effet à la forme phosphorylée de l’activateur de transcription CREB (CRE Binding protein) qui reconnaît spécifiquement les éléments CRE. CBP est un coactivateur de transcription et son altération peut influer sur l’expression des différents gènes cibles de CREB ou d’autres facteurs de transcription qui interagissent avec lui, tels que TFIIB, c-Jun, c-Fos, c-Myb, des récepteurs nucléaires, et la protéine P/CAF qui présente une activité acétylase d’histone. CBP interagit aussi avec les protéines SREBP1 et 2 (sterol regulatory element binding proteins) qui activent la transcription de gènes dont les produits interviennent dans l’internalisation cellulaire (récepteurs des LDL) ou la synthèse du cholestérol (HMG-CoA synthase et HMG-CoA réductase), ce qui indique que CBP régule une partie du métabolisme du cholestérol. Nous avons développé la recherche de mutations dans le gène CBP chez les patients par des techniques de génétique moléculaire (DHPLC, PCR-séquence, QMF-PCR) et la recherche de petits remaniements génomiques par technique d’hybridation génomique comparative sur microréseaux d’ADN (CGH-array) ou puces à ADN [20].
Le gène CBP a été impliqué dans des leucémies aiguës myéloïdes (sous-types M4 et M5) par des mécanismes de translocations chromosomiques, suggérant que CBP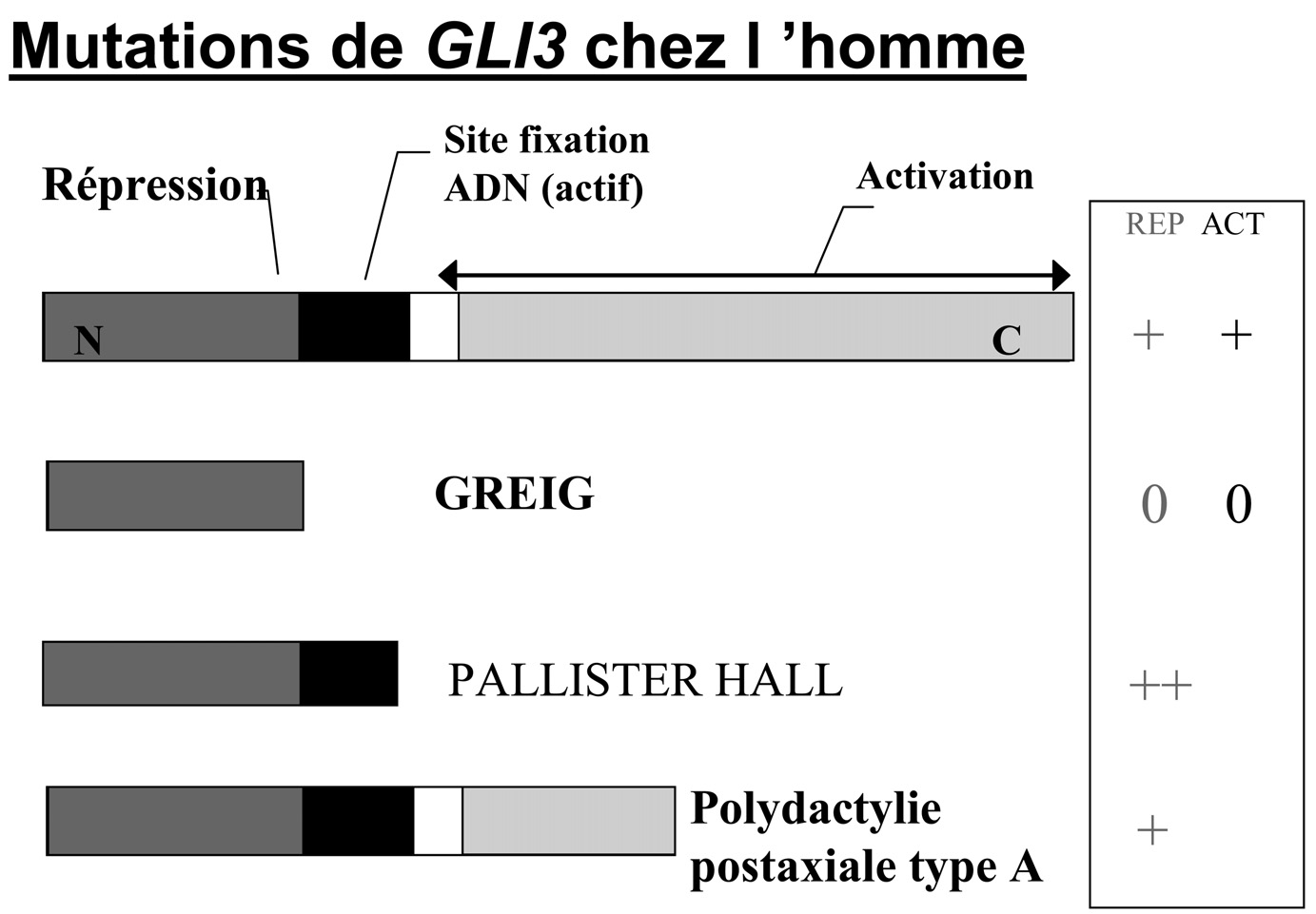
Fig. 5. — Mutations du gène GLI3 et anomalies du développement (syndrome de Greig, syndrome de Pallister-Hall, polydactylie post-axiale) chez l’homme selon le mécanisme de répression (REP), d’activation (ACT) ou des deux.
pouvait jouer un rôle dans la leucémogenèse. Un mécanisme de perte de l’hétérozygotie pourrait être impliqué. CBP et CREB sont d’importants facteurs de régulation, mais le mécanisme par lequel un déficit du niveau d’expression de CBP affecte le développement reste peu clair. Chez la Drosophile, CBP fonctionne comme un coactivateur de cubitus interruptus , un composant de la voie de signalisation de hedgehog. Cette voie de transduction du signal a un rôle important dans le développement, et cela pourrait expliquer la contribution de CBP dans la formation embryonnaire chez les vertébrés. Le SRT est un exemple de syndrome malformatif avec retard mental dû à une anomalie généralisée du contrôle de l’expression des gènes.
AUTRES SYNDROMES ET FACTEURS DE TRANSCRIPTION
Parmi les craniosténoses dont la physiopathogénie moléculaire a récemment explosé notamment avec les gènes de la famille FGFR (Fibroblast Growth Factor Receptor), on peut noter le syndrome de Saethre-Chotzen dû à des mutations du gène TWIST [21]. Ce gène code pour un facteur de transcription de la famille basique hélice-boucle-hélice et joue un rôle fondamental dans la gastrulation.
Un intérêt récent s’est porté sur la diversité des phénotypes associés à différentes mutations dans le gène à doigt de zinc GLI3 qui comprend trois domaines : un site
Fig. 6. — Hedgehogopathies dues à des mutations sur la voie de transduction du signal médiée par Sonic Hedgehog Légende → : activation T : répression Hh : Hedgehog (Sonic Hh chez l’homme) PTC1 : gène codant pour la protéine Patched Smo : gène smoothened Fused : gène Fused Su(Fu) : gène Suppressor of fused Cos2 : gène costal 2 GLI3 : gène codant pour un facteur de transcription initialement identifié dans le glioblastome CBP : gène codant pour la protéine CREBBP (CREB Binding Protein) Wnt : voie de signalisation Wint TGFβ : Transforming Growth Factor β de liaison à l’ADN, un domaine répresseur et un domaine activateur. Le spectre clinique va de la polydactylie isolée (post-axiale type A ou B ou pré-axiale), à une forme associée à des anomalies modérées (macrocéphalie et polydactylie du syndrome de Greig) ou plus sévères (hamartome de l’hypothalamus, dysmorphie oro-faciale, luette bifide, imperforation anale dans le syndrome de Pallister-Hall [22-24]. Dans le syndrome de Greig, la mutation est situé dans la partie proximale du gène GLI3, entrainant la perte des activités de répression et d’activation du gène
Table 2. — Mutations des gènes FOX en pathologie humaine Syndrome
Gène
Glaucome congénital FOXC1 Agénésie thyroïdienne FOXE1 Anomalie de chambre antérieure de l’œil avec cataracte FOXE3 Lymphoedème-distichiasis FOXC2 BPES FOXL2 IPEX (polyendocrinopathie et entéropathie liée à l’X FOXP3 Trouble mixte du langage et dyspraxie verbale FOXP2 Cardiomyopathie dilatée et troubles psychiatriques FOXD4 Anencéphalie et craniorachischisis FOXN1 d’où le phénotype intermédiaire. Dans le syndrome de Pallister-Hall, la mutation se situe dans le domaine de fixation à l’ADN ce qui entraîne le phénotype le plus sévère, l’effet de répression étant activé par la localisation de la mutation qui a un effet d’activation de l’effet répresseur. Dans la polydactylie post-axiale (rayon cubital), la mutation se situe dans le domaine activateur dans la partie C-terminale, avec un effet de répression non affecté, mais restant modéré (non activé) car le site de liaison à l’ADN n’est pas touché (Fig. 5). Ce type de corrélation phénotype/génotype peut s’avérer utile dans le cadre du conseil génétique.
Le gène GLI3 est un facteur de transcription impliqué dans la voie de transduction du signal médié par Sonic Hedgehog (Shh) à travers les récepteurs patched et smoothened et via des cibles nucléaires comme les BMP (Bone Morphogenetic Proteins). On connaît maintenant les pathologies associées à des mutations dans certains de ces gènes impliqués dans la même voie de signalisation cellulaire (Fig. 6).
Des critères cliniques identiques peuvent se retrouver dans certains cas liés à des anomalies dans des gènes différents mais impliqués dans la même voie de transduction.
La voie de Shh est connue pour son rôle dans le développement embryonnaire ; CBP impliqué dans le déterminisme du syndrome de Rubinstein-Taybi, ZIC3 lié aux anomalies de latéralisation type situs [25], et SALL1 (SAL-Like 1) à l’origine du syndrome de Townes-Brocks [26] interagissant également dans la voie de transduction médiée par Shh. Le gène SALL1 est un homologue de la famille des gènes Spalt (Sal) de la Drosophile et intervient dans la différenciation postérieure céphalique et posté- rieure caudale.
D’autres exemples peuvent être cités comme les gènes Sox (SRY-Box ou SRY-Related HMG-Box) avec des mutations de Sox-9 dans la dysplasie campomélique [27], ou encore des mutations du facteur de transcription spécifique des ostéoblastes OSF2/CBFA1 (Osteoblast Specific Transcription Factor/Core-Binding Factor
Alpha Subunit 1) dans la dysplasie ou dysostose cleido-crânienne [28]. De nombreuses pathologies ont été associées à des mutations dans des gènes de la famille FOX (Forkhead Box) (Table 2).
CONCLUSION
Les facteurs de transcription sont probablement pour un grand nombre des agents majeurs du développement embryonnaire humain. Ils se caractérisent par des séquences très conservées parmi les espèces animales et par des motifs spécifiques de liaison à l’ADN. Ils ont une fonction de régulation de l’expression d’autres gènes, permettant un développement embryonnaire normal en l’absence de mutation sur ces gènes. La génétique clinique et l’identification des phénotypes dus à des mutations dans des gènes codant pour des facteurs de transcription nous permettent de mieux comprendre la fonction de ces gènes et les processus du développement normal et anormal de l’être humain. Cette approche permet de mieux appréhender les problè- mes diagnostiques à visée de prise en charge des enfants atteints et de conseil génétique pour les familles [29].
BIBLIOGRAPHIE [1] Nüsslein-Volard C., Wieschaus E. — Mutations affecting segment number and polarity in drosophila. Nature , 1980, 287 , 795-801.
[2] Engelkamp D., Van Heyningen V. — Transcription factors in disease.
Hum. Mol. Genet. , 1996, 6 , 334-42.
[3] Mann RS., Affolter M. — Hox proteins meet more partners.
Curr. Op. Genet. Dev ., 1998, 8 , 423-9.
[4] Muragaki Y., Mundlos S., Upton J., Olsen B.R. — Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science , 1996, 272 , 548-51.
[5] Kondo T., Zacharry J., Innis J.W., Duboule D. — Of fingers, toes and penises.
Nature 1997, 390 , 29.
[6] Johnson D., Kan S., Oldridge M., Trembath R.C., Roche P., Esnouf R.M., et al —
Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E. Am. J. Hum. Genet. , 2003, 72 , 984-997.
[7] Mortlock D.P., Innis J.W. — Mutation of HOXA13 in hand-foot-genital syndrome.
Nature
Genet, 1997, 15, 179-80.
[8] Jabs E.W., Muller U., Li X. et al . — A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis.
Cell , 1993, 75 , 443-50.
[9] Vastardis H., Karimbux N., Guthua S.W. et al . — A human MSX1 homeodomain missense mutation causes selective tooth agenesis.
Nature Genet, 1996, 13 , 417-21.
[10] Brunelli S., Faiella A., Capra V. et al . — Germline mutations in the homeobox gene EMX2 in patients with severe schizencephaly.
Nature Genet , 1996, 12 , 94-6.
[11] Li Q.Y., Newbury-Ecob R.A., Terrett J.A. et al . — Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family.
Nature Genet, 1997, 15 , 21-9.
[12] Bamshad M., Lin R.C., Law D.J. et al . — Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome.
Nature Genet , 1997, 16 , 311-5.
[13] Zweier C., Sticht H., Aydin-Yaylagül I., Campbell C.E., Rauch A. — Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions.
Am. J. Hum. Genet. , 2007, 80 , 510-7.
[14] Attié-Bitach T., Lyonnet S., Vekemans M., Lacombe D. — Gènes PAX et anomalies du développement. MT Pédiatrie 1998, 1 , 517-26.
[15] Barr F.G., Galili N., Holick J., Biegel J.A., Rovera G., Emanuel B.S. — Rearrangement of the PAX-3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nature Genet. , 1993, 3 , 113-7.
[16] Tassabehji M., Read A.P., Newton V.E. et al . — Waardenburg syndrome patients have mutations in the human homologue of the Pax-3 paired box gene.
Nature , 1992, 355 , 635-6.
[17] Tassabehji M., Newton V.E., Read A.P. — Waardenburg syndrome type 2 caused by mutations in the human microphtalmia (MITF) gene. Nature Genet. , 1994, 8 , 251-5.
[18] Taine L., Goizet C., Wen Z.Q. et al . — Submicroscopic deletion of chromosome 16p13.3 in patients with Rubinstein-Taybi syndrome.
Am. J. Med. Genet ., 1998, 78 , 267-70.
[19] Petrij F., Giles R.H., Dauwerse H.G. et al . — Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP.
Nature 1995, 376 , 348-51.
[20] Stef M., Simon D., Burgelin I., Guisle I., Chevalier C., Delrue M.A., et al . — Testing and improving experimental parameters for the use of low molecular weight targets an array-CGH experiments. Hum. Mut ., 2006, 27 , 1143-1150.
[21] El Ghouzzi V.E., Le Merrer M., Perrin-Schmitt F. et al . — Mutations of the TWIST gene in the Saethre-Chotzen syndrome.
Nature Genet., 1997, 15 , 42-6.
[22] Wild A., Kalff-Suske M., Vortkamp A. et al . — Point mutations in human GLI3 cause Greig syndrome.
Hum. Mol. Genet., 1997, 6 , 1979-84.
[23] Kang S., Graham Jr J.M., Olney A.H., Biesecker L.G. — GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nature Genet. , 1997, 15 , 266-8.
[24] Radhakhrishna V., Wild A., Grzeschik K.H., Antonorakis SE. — Mutation in GLI3 in post-axial polydactyly type A. Nature Genet. , 1997, 17 , 259-60.
[25] Gebbia M., Ferrero G.B., Pilia M.T. et al . — X-linked situs abnormalities result from mutations in ZIC3.
Nature Genet. , 1997, 17 , 305-8.
[26] Kohlhase J., Wischermann A., Reichenbach H. et al . — Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome.
Nature Genet. , 1998, 18 , 81-3.
[27] Foster J.W., Dominguez-Steglich M.A., Guloli S. et al . — Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene.
Nature 1994, 372 , 525-30.
[28] Lee B., Thirunavukkarasu K., Zhou L. et al . — Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia.
Nature Genet. , 1997, 16 , 307-10.
[29] Battin J., Lacombe D. — La dysmorphologie et la syndromologie, une activité de généticiens pédiatres (Editorial). Arch Pédiatr , 1995, 7 , 615-18.
DISCUSSION
M. Pierre CORVOL
Les gènes du développement sont-ils définitivement éteints à l’âge adulte ou peuvent-ils être réexprimés dans certains cas, par exemple en cas de plasticité tissulaire (neurale ou autre) ?
Certains gènes du développement sont éteints après la phase d’expression embryonnaire.
D’autres continuent effectivement d’être exprimés durant la vie adulte, comme certains gènes codant pour des facteurs de transcription (tel CREBBP), ce qui peut intervenir dans la plasticité neuronale.
M. Christian NEZELOF
Ces phénomènes doivent être vus de façon dynamique. Comment peut-on expliquer les différences phénotypiques accompagnant la mue survenant des insectes qui passent en quelques heures, de la larve à la chenille, au papillon : modifications qui existaient sans modification du DNA germinal ?
L’évolution morphologique des espèces animales au cours de leur vie est la plus marquée chez les insectes avec les différentes métamorphoses, mais cela peut être rapproché de la période pubertaire dans l’espèce humaine. Ces modifications sont sous la dépendance d’hormones et cela est aussi le cas chez les insectes (ecdysone…), mais cette production hormonale est contrôlée par l’expression de gènes qui s’expriment à un moment spécifique de la vie. Certains gènes impliqués dans ces contrôles morphogénétiques chez l’insecte commencent à être identifiés. Cela peut aussi être rapproché des gènes d’horloge comme ceux qui règulent le cycle nycthéméral de sécrétion de mélatonine impliquée dans le sommeil.
M. Jean COSTENTIN
La phocomélie induite par le thalidomide procède-t-elle d’un effet génotoxique ?
La thalidomide est un agent tératogène responsable essentiellement de malformations des membres chez l’embryon lorsqu’il est pris par la femme enceinte à une période critique du développement. Certains agents tératogènes perturbent en effet l’expression de gènes du développement ou de voies de signalisation cellulaire. Les hypothèses actuelles vont dans ce sens pour la thalidomide. Par ailleurs, les gènes impliqués dans le déterminisme des phocomélies sont aujourd’hui mal connues, contrairement à ceux des ectrodactylies.
M. Jacques BATTIN
Si l’on prend la famille HOX qui contrôle le développement des nageoires, des pattes et des mains de l’homme, ces gênes sont conservés mais aboutissent à des résultats bien différents.
Comment l’expliquer ?
L’évolution phylogénétique des huit gènes Hom-c de Drosophila Melanogaster (Antennapedia, Bithorax) s’est faite vers trente-neuf gènes HOX répartis en quatre groupements de gènes ou clusters dans l’espèce humaine. On peut en effet penser que cette diversité participe de la complexité morphologique, à savoir qu’il est plus complexe de façon caricaturale de structurer les membres de l’homme que les ailes de la mouche.
M. Raymond ARDAILLOU
Peut-on réaliser des mutations conditionnelles dans le temps chez les insectes comme on le fait chez la souris de manière à bloquer la métamorphose ?
Il est tout à fait possible d’envisager des expérimentations de mutagenèse dirigée (type mutants « knock-out » ou K.O.) chez les insectes comme la drosophile, de la même manière que cela est réalisé chez la souris par exemple. Certaines de ces mutations homéotiques peuvent être des modèles pour des anomalies du développement d’origine génétique.
* Génétique Médicale, Hôpital Pellegrin-Enfants, CHU de Bordeaux ; Laboratoire de Génétique Humaine, Université Victor Segalen Bordeaux 2, 33076 Bordeaux. Tirés à part : Professeur Didier Lacombe, même adresse Article reçu le 9 juillet 2008, accepté le 6 novembre 2008
Bull. Acad. Natle Méd., 2009, 193, no 4, 931-945, séance du 28 avril 2009