
Résumé
Les lésions de la maladie d’Alzheimer sont des accumulations protéiques et des pertes cellulaires et synaptiques. Les premières permettent le diagnostic, les secondes sont non spécifiques. Le peptide A β s’accumule dans le milieu extracellulaire, la protéine dans les neurones avec une chronologie et une topographie distinctes. Les lésions riches en peptide A β , intéressent d’abord le néocortex, et celles dues à l’accumulation de protéine tau, la région hippocampique ; les premières se constituent rapidement, les secondes se développent par stades stéréotypés, affectant successivement les aires associatives multi et unimodales, puis primaires. Les mutations de l’APP, le précurseur du peptide A β , sont à l’origine de maladie d’Alzheimer familiale autosomique dominante. Des dépôts de peptide A β apparaissent dans des lignées transgéniques, portant le gène muté de l’APP (souris APP). L’immunothérapie par le peptide A β prévient chez la souris APP l’apparition des lésions. Tentée chez l’homme, elle a été interrompue parce qu’elle provoquait une méningoencéphalite. De nouvelles tentatives devraient permettre de déterminer si la prévention des dépôts permet de prévenir aussi la cascade de réactions qui conduit aux symptômes .
Summary
The brain lesions associated with Alzheimer’s disease are caused by extracellular accumulation of A β peptide and intracellular accumulation of tau protein. A β peptide makes the core of the senile plaque (the ‘‘ focal deposit ’’) ; it is also present in the extracellular ‘‘ diffuse deposits ’’ and in the vessel walls. Neurofibrillary tangles, and neuropil threads are composed of hyperphosphorylated tau that also accumulates in the processes of the corona of the senile plaque. The A β deposits first involve the neocortex, while the tau pathology is initially found in the hippocampal region. A β deposits first occur in the neocortex, while intracellular tau accumulation mainly affect the hippocampal region. A β peptide deposits are initially found in all the neocortical areas, then involve the hippocampus and the subcortical nuclei. Tau lesions successively involve the hippocampal regions, multi- and uni-modal areas and finally the primary cortices in stereotyped stages. Mutations of APP, the precursor of A β peptide, cause autosomal dominant familial Alzheimer disease, suggesting that a cascade of reactions link A β overproduction, tau pathology and the clinical phenotype. Transgenic mice bearing the mutated human APP gene (APP mice) develop A β deposits. Systemic injection of A β peptide prevents the deposition of A β peptide. However, a clinical trial had to be interrupted when meningoencephalitis occurred in a significant proportion of treated patients. Post mortem studies showed a relative scarcity of A β deposits. Forthcoming immunotherapy studies should soon show whether the prevention of A β deposition interrupts disease progression .
INTRODUCTION
Les mutations de l’APP (Amyloid Precursor Protein), le précurseur du peptide Aβ, sont à l’origine de maladie d’Alzheimer familiale autosomique dominante — ce qui laisse supposer qu’il existe une cascade de réactions entre la production accrue de peptide Aβ et l’apparition de protéine tau. L’immunothérapie par le peptide Aβ prévient chez la souris transgénique APP l’apparition des lésions. Tentée chez l’homme, elle a été interrompue parce qu’elle provoquait une leucoencéphalite. Les dépôts parenchymateux de peptide Aβ étaient réduits dans les quelques cas humains traités, qui ont pu être examinés post-mortem. De nouvelles tentatives devraient permettre de déterminer si la prévention des dépôts permet d’interrompre la cascade de réactions qui conduit aux symptômes.
Le neuropathologiste, comme le neurologue, se base sur deux éléments pour porter un diagnostic : la topographie des lésions et leur nature. La topographie était naguère essentielle. Le terme de maladie de Pick, par exemple, était pratiquement synonyme de démence à lésions frontales. Aujourd’hui, par un retour de balancier, le diagnostic repose presque exclusivement sur l’analyse de la nature des lésions.
Celles-ci, à leur tour, sont de deux types [1]. Elles peuvent être non spécifiques : ce sont, par exemple, la perte neuronale et la gliose, — véritable voie finale commune des maladies neurodégénératives. Elles peuvent au contraire être spécifiques (ou considérées comme telles aujourd’hui) : ce sont elles qui permettent d’identifier la maladie. Ces lésions spécifiques sont presque toujours des accumulations de protéines, extracellulaires (ce sont les dépôts), intracellulaires (les inclusions).
Les inclusions et les dépôts se prêtent à l’analyse biochimique car ils sont constitués d’une protéine majoritaire. Le nom de la protéine accumulée peut être utilisé pour désigner un groupe d’affections, comme dans les termes ‘‘ tauopathie ’’ ou ‘‘ synucléinopathie ’’. Le succès, au moins expérimental, de l’immunothérapie par l’une des
protéines impliquées dans la maladie d’Alzheimer montre que les accumulations protéiques jouent un rôle essentiel dans la physiopathologie.
Neuropathologie
La diminution du poids du cerveau peut être considérable. L’atrophie porte préfé- rentiellement sur le pôle temporal, l’hippocampe, l’amygdale [2], c’est-à-dire les régions où, comme nous le verrons, la pathologie neurofibrillaire est la plus marquée.
L’étude de la maladie d’Alzheimer a été rendue possible par la mise en évidence des lésions au moyen de techniques argentiques, développées à la fin du XIXe et au début du XXe siècle à partir des méthodes utilisées pour la confection des miroirs et la chimie de la photographie. L’immunohistochimie a aujourd’hui presque complètement remplacé les techniques argentiques car elle permet non seulement de révéler l’accumulation protéique mais aussi d’en déterminer la nature grâce à l’utilisation d’anticorps spécifiques.
La nature des lésions
On peut classer les lésions en deux groupes, par excès et par manque : les premières sont principalement dues à des accumulations protéiques . Les deux principales sont les plaques séniles et les dégénérescences neurofibrillaires qui permettent le diagnostic. Les secondes — perte de neurones et de synapses — ont une grande importance physiopathologique mais elles ne sont ni spécifiques ni faciles à objectiver. On peut classer les lésions par excès selon les protéines qui s’y accumulent : peptide Aβ ou protéine tau — et c’est l’option que nous avons choisie ici bien que la plaque sénile, composite, comporte les deux protéines.
Les lésions par excès
L’accumulation extracellulaire de peptide A β chez l’homme et la souris transgénique
APP.
Chez l’homme Le peptide A4, amyloïde ou Aβ, est normalement produit dans le cerveau. Il provient de l’APP, son précurseur transmembranaire, à la suite de deux coupures séquentielles dues aux activités enzymatiques β- et γ- secrétases. Des mutations de l’APP ou de la préséniline, un des composants de la γ-secrétase, sont associées à des maladies d’Alzheimer familiale. Il existe de multiples isoformes de peptide Aβ. Du côté carboxy-terminal, il se termine parfois à l’acide aminé 42 (Aβ 42) ; parfois, plus court, il finit à l’acide aminé 40 (Aβ 40). Du coté amino-terminal, la spectrométrie de masse a montré que le peptide Aβ pouvait être tronqué.
Dans la maladie d’Alzheimer, le peptide s’accumule en dehors des cellules et échappe aux divers systèmes, enzymatiques ou cellulaires, d’élimination. Il s’enrichit en feuillets β-plissés et, devenu insoluble, précipite sous forme amyloïde dans le tissu nerveux. Le ‘‘ dépôt focal ’’ de peptide Aβ [3] peut former le cœur amyloïde de la plaque sénile qui comporte également une couronne faite de prolongements nerveux. Le ‘‘ dépôt diffus ’’ du même peptide Aβ (fig. 1) n’est pas amyloïde. Il est de plus grande taille (pouvant atteindre quelques centaines de microns), moins concentré et souvent mal limité. Il n’a pas de composante nerveuse. Les dendrites ou les axones inclus dans le dépôt diffus sont certes modifiés mais ces anomalies n’ont pas de retentissement majeur car des dépôts diffus peuvent être trouvés, parfois en grand nombre, chez des personnes âgées dépourvues de troubles intellectuels ou chez lesquelles les altérations cognitives sont minimes [4]. Dans certaines régions céré- brales, comme le striatum ou le cervelet, les dépôts sont toujours diffus et ne forment jamais une plaque sénile typique comportant un cœur amyloïde et une couronne nerveuse. Dans d’autres régions, au contraire, le dépôt diffus de peptide A β pourrait constituer le stade précoce de la plaque sénile.
Le peptide Aβ peut aussi s’accumuler dans la paroi des vaisseaux, souvent de façon segmentaire (seule une partie du vaisseau est touché) ou sur une partie seulement de sa circonférence, constituant l’« angiopathie amyloïde ». Les petites artères perforantes du cortex cérébral sont le plus souvent intéressées mais des artères méningées plus volumineuses, dans certains cas des capillaires, et peut-être des veines peuvent l’être aussi. Les termes de micro et de macro-angiopathie sont utilisés aujourd’hui pour décrire l’atteinte des petites ou des grosses artères. L’angiopathie amyloïde est, sinon constante, du moins fréquente dans la maladie d’Alzheimer sporadique [5]. La microangiopathie est liée aux lésions neurofibrillaires de la maladie d’Alzheimer alors que la macroangiopathie ne l’est pas [6]. L’angiopathie amyloïde est particulièrement sévère dans certaines mutations de l’APP ou de la préséniline 1. C’est ainsi que la maladie vasculaire est au premier plan dans la mutation dite Hollandaise qui remplace la glutamine en position 693 de l’APP par l’acide glutamique [7].
La composition des dépôts d’Aβ varie selon leur localisation : les dépôts diffus sont principalement constitués de peptide Aβ 42, les dépôts vasculaires comportent principalement de l’Aβ 40 et le cœur des plaques séniles comporte les deux formes du peptide.
Chez les souris transgéniques L’introduction, dans des lignées de souris, du gène muté de l’APP, seul (souris APP) ou associé au gène muté de la préséniline 1 (APPxPS1), l’un des composants de la γ-secrétase, provoque l’apparition de dépôts, diffus et focaux, de peptide Aβ, associés à une réaction microgliale, semblables à ceux qui sont observés chez l’homme [8]. Lorque le gène de la préséniline 1 humaine mutée remplace le gène murin et qu’il est donc placé sous le contrôle du promoteur de la souris (souris APPxPS1 pour ki knocked in), on observe, outre les dépôts, une perte neuronale localisée dans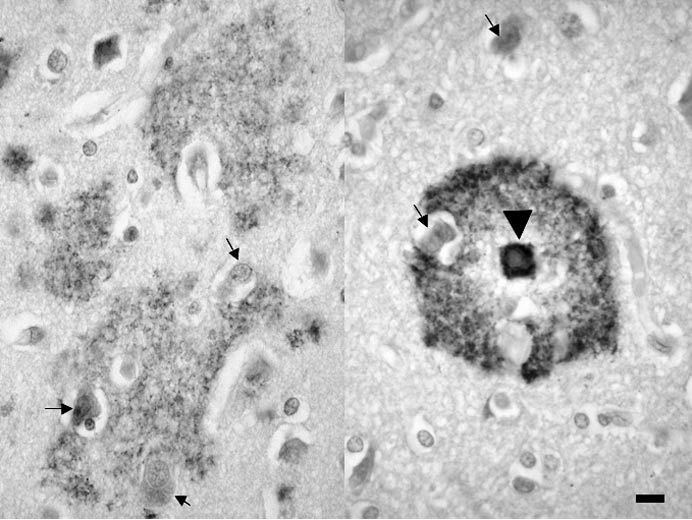
FIG. 1. — Dépôts diffus et focaux de peptide Aβ.
Dépôt diffus (à gauche) et focal (pointe de flèche à droite) de peptide Aβ.
Quelques neurones sont indiqués par des flèches noires. Les dépôts apparaissent en gris sombre ou noir. La barre d’échelle correspond à une longueur de 20 µm.
Technique : Isocortex cérébral. Prélèvement post mortem. Coupe à la paraffine. Anticorps primaire :
anti-corps monoclonal de souris dirigé contre l’épitope 8-17 du peptide Aβ. Clone 6F/3D (Dako, Glostrup). Révélation par la méthode du complexe Avidine-Biotine Peroxydase. Chromogène :
diaminobenzidine.
le secteur CA1 de l’hippocampe [9], ce qui rapproche ce modèle de la pathologie humaine. Comme nous le verrons plus loin, les souris APP ou APPxPS1 reproduisent cependant très imparfaitement la pathologie neurofibrillaire.
L’accumulation intracellulaire de protéine tau
Alois Alzheimer découvrit, grâce aux techniques argentiques, la ‘‘ Fibrillenveränderung ’’, la dégénérescence neurofibrillaire (DNF). En microscopie électronique, la DNF est constituée d’une paire de filaments appariés en hélice (paired helical filaments, PHF [10]). L’adjectif fibrillaire accolé au terme de dégénérescence est dû, en fait, à une erreur d’interprétation : les ‘‘ neurofibrilles ’’ ont été vues dans les neurones dès la fin du XIXe siècle. Les progrès de la microscopie électronique et de la biologie cellulaire ont montré qu’il s’agissait des ‘‘ neurofilaments ’’, le filament intermédiaire du cytosquelette neuronal. Mais la ‘‘ dégénérescence neurofibrillaire ’’ (« DNF ») n’est pas constituée de neurofilaments, comme on l’a d’abord cru
mais d’une protéine normale qui intervient dans la polymérisation des neurotubules, la protéine tau [11] isolée en 1977 et séquencée plus récemment [12]. Six isoformes sont connues. La protéine comporte des segments répétitifs au nombre de 3 (tau 3R) ou de 4 (tau 4R). Les lésions de la maladie d’Alzheimer comportent à la fois des formes 3R et 4R, les unes et les autres anormalement phosphorylées [13]. Les mutations de la protéine tau ne sont pas associées à la maladie d’Alzheimer mais à des démences fronto-temporales. L’introduction du gène muté de la protéine tau dans des lignées transgéniques peut, si le promoteur est adéquat, provoquer l’apparition de dégénérescences neurofibrillaires dans l’isocortex et le cortex limbique, mais celles-ci ne sont pas associées à des dépôts de peptide Aβ.
Au cours de la maladie d’Alzheimer, la protéine tau s’accumule exclusivement dans le neurone dont elle occupe les différents compartiments : dans le corps cellulaire, les amas de protéine tau constituent la dégénérescence neurofibrillaire ; ils sont aussi présents dans les extrémités axonales qui entourent le cœur de la plaque sénile et constituent sa « couronne » ; les « filaments du neuropile » (neuropil threads) appelés aussi, en français, « fibres tortueuses » [14], sont principalement en rapport avec l’accumulation de protéine tau dans les dendrites [15] (fig. 3). Enfin, la concentration de la protéine tau peut être élevée dans le cytoplasme du neurone, sans que l’agrégation ne se soit encore produite : le marquage du corps cellulaire et des prolongements nerveux par les anticorps dirigés contre la protéine tau est alors désigné par le terme de pré-DNF.
La plaque sénile : un cœur de peptide A β et une couronne de protéine tau
La plaque sénile « classique » ou « mûre » est constituée d’un dépôt focal (fig. 1 et 2) amyloïde de peptide Aβ et d’une couronne faite d’un entrelacs de prolongements axonaux chargés de protéine tau ou, pour certains d’entre eux, d’APP. Nombre de ces prolongements sont aussi marqués par les anticorps anti-ubiquitine, ce qui indique que la voie de dégradation protéique du protéasome est activée [16]. La plaque sénile classique comporte invariablement une ou plusieurs cellules microgliales activées [17] — fig 2. Leur rôle a été discuté : pour certains, elles sont indispensables à la transformation amyloïde du peptide Aβ ; pour d’autres, elles phagocytent les fibrilles d’amyloïde déjà produits. Quoiqu’il en soit, elles expriment des chimiokines, contribuant à entretenir une inflammation locale, à bas bruit, sans composante lymphocytaire. Cette réaction inflammatoire comporte des composants pré- coces de la cascade du complément [18], de l’alpha 1 anti-chymotrypsine et des cytokines proinflammatoires [19]. Les expériences de « vaccination » par des préparations comportant des épitopes du peptide Aβ administrés de façon systémique, indiquent que la microglie activée peut, dans certaines conditions, éliminer la composant amyloïde de la plaque sénile [20]. La plaque comporte également du cholestérol et de l’apolipoprotéine E, le transporteur du cholestérol [21].
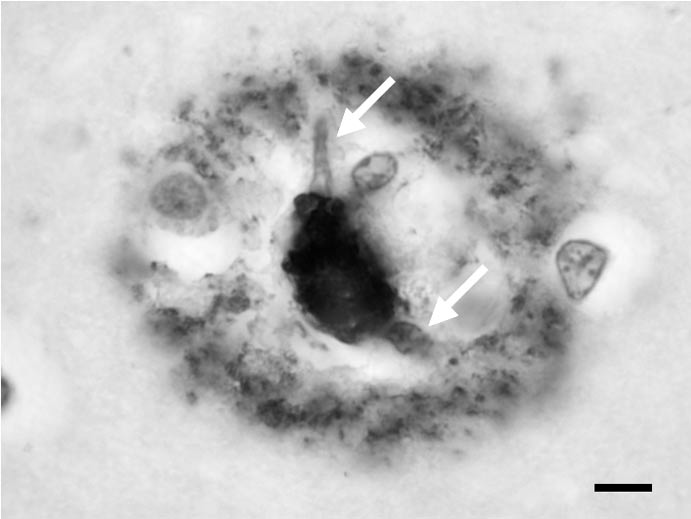
FIG. 2. — Les macrophages dans la plaque sénile.
Les noyaux des macrophages sont indiqués par des flèches blanches. Ils sont situés dans une couronne dépourvue d’immunoréactivité qui entoure le dépôt focal de peptide Aβ. C’est l’activation du macrophage qui est supposée permettre la résorption des dépôts au cours de l’immunothérapie par le peptide Aβ.
Barre d’échelle = 20 µm. Isocortex cérébral. Prélèvement post mortem. Coupe à la paraffine.
Anticorps primaire : anti-corps monoclonal de souris dirigé contre l’épitope 8-17 du peptide Aβ.
Clone 6F/3D (Dako, Glostrup). Révélation par la méthode du complexe Avidine-Biotine Peroxydase. Chromogène : diaminobenzidine.
La couronne de la plaque sénile est principalement constituée de prolongements axonaux. Les neurones d’origine de ces axones sont encore imparfaitement connus.
Certains sont catécholaminergiques, mais beaucoup paraissent issus des connexions cortico-corticales [22]. Chez la souris transgénique, les axones qui entourent les dépôts de peptide Aβ proviennent en majorité de connexions cortico-corticales [23].
La couronne nerveuse de la plaque sénile se développe après le dépôt de peptide Aβ [24]. Chez la souris transgénique APP ou APPxPS1, les dépôts de ce peptide sont entourés de prolongements nerveux élargis, enrichis en protéine tau hyperphosphorylée [8], mais d’authentiques DNF ou PHF ne sont pas observées. La connexion entre les pathologies Aβ et tau ne peut donc être reproduite aujourd’hui dans ce modèle animal — le plus intéressant et le plus utilisé — de la maladie.
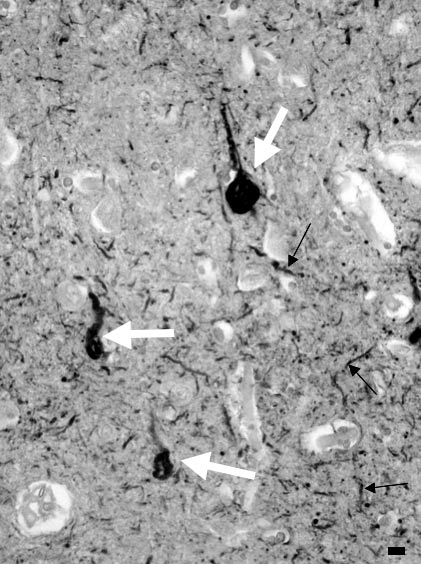
FIG. 3. — Pathologie neurofibrillaire révélée par l’anticorps anti-tau.
Toutes les structures colorées en marron sont pathologiques. Elles comportent des dégénérescences neurofibrillaires, accumulation de tau dans le corps cellulaire des neurones (flèches blanches volumineuses) et les fibres tortueuses (« neuropil threads »), dont certaines sont marquées par les petites flèches noires.
Barre d’échelle = 20 mm. Technique : Isocortex cérébral. Prélèvement post mortem. Coupe à la paraffine. Anticorps primaire : anticorps polyclonal de lapin dirigé contre la partie C-terminale de la protéine tau (exprimée chez E. Coli ), Dako, Glostrup. Complexe Avidine-Biotine Peroxydase.
Chromogène : diaminobenzidine.
Les lésions par manque
La perte neuronale
Son ampleur est très diversement appréciée. Le compte non corrigé des corps cellulaires neuronaux sur une coupe examinée au microscope est biaisé : il surestime en effet les grands neurones (qui ont une probabilité plus grande d’être coupés) par rapport aux petits. Pour palier ce biais, la technique dite du ‘‘ disecteur ’’ (deux sections d’où l’orthographe différente de celle de dissection) a été la plus utilisée. Elle ne comptabilise que les profils neuronaux présents sur l’une des coupes et absents de l’autre — ce qui revient à dénombrer le pôle inférieur de la cellule, un point géométrique de dimension nulle qui n’est pas susceptible d’être recoupé.
L’importance physiopathologique de la mort neuronale fait également l’objet de discussions : pour certains auteurs, elle constitue un élément essentiel et précoce de la physiopathologie, à l’origine directe des symptômes [25]. Pour d’autres, au contraire, elle survient tardivement, à une phase de la maladie où les symptômes sont déjà installés [26]. Les premières études mentionnaient une perte sélective des grands neurones [27], qui, à la lumière de la stéréologie, peut être interprétée comme la conséquence tout aussi bien d’une diminution de leur volume, c’est-à-dire de leur atrophie, que d’une mort cellulaire [28]. La technique du disecteur a permis de montrer qu’il n’existait pas de ‘‘ perte neuronale néocorticale globale ’’ dans la maladie d’Alzheimer [29]. En revanche, la méthode des polygones de Voronoï, qui permet d’analyser les variations locales de densité cellulaire, a montré que cette perte est très marquée dans certaines aires corticales (comme l’aire entorhinale) ou dans certaines couches. Elle prédomine dans les couches III et V, celles précisément qui sont affectées par les DNF [26]. La perte neuronale est, de plus, statistiquement liée à leur densité. L’analyse multivariée montre cependant que le déficit intellectuel est lié plus à la densité des DNF qu’à la perte neuronale [26]. Celle-ci est plus marquée que ne le voudrait le nombre des dégénérescences neurofibrillaires qui pourraient donc ne pas être responsables, ou seules responsables, de la mort neuronale [25].
La perte des synapses
Son rôle physiopathologique a été jugé central par quelques auteurs [30]. La microscopie électronique a révélé une diminution de la densité des synapses mais une préservation de leur surface d’apposition — ce qui suggère que la taille des synapses augmente tandis que leur nombre diminue [31]. L’immunohistochimie de la synaptophysine, protéine vésiculaire présynaptique, révèle une diminution importante de l’immunoréactivité, interprétée, peut-être rapidement, comme la consé- quence de la perte des synapses [32]. La baisse varie en fait selon les marqueurs utilisés et ne semble pas liée à l’accumulation de peptide Aβ [33]. Elle n’est sans doute pas le meilleur corrélat des troubles cognitifs [34]. Dans cette dernière étude, c’était à nouveau la densité des dégénérescences neurofibrillaires qui était la plus étroitement liée à la démence.
Topographie des lésions
Les lésions de la maladie d’Alzheimer, pratiquement confinées à la substance grise, ont une répartition remarquablement hétérogène ; elle est différente pour le peptide Aβ et la protéine tau. Les dépôts de peptide Aβ, diffus ou focaux, siègent principalement dans l’isocortex ; des dépôts uniquement diffus sont constatés dans le striatum, le thalamus, le tronc cérébral et le cervelet à des stades plus tardifs [35].
Les cellules pyramidales des champs ammoniens de l’hippocampe, de la couche II du cortex entorhinal et de la couche III de l’isocortex, les cellules multipolaires du noyau basal de Meynert cholinergique sont celles qui sont le plus précocement et le
plus souvent affectées par la pathologie neurofibrillaire. Au contraire, les cellules granulaires du gyrus denté et de la couche IV de l’isocortex ne sont affectées que rarement et tardivement. Les stades de Braak décrivent la topographie des lésions neurofibrillaires [36] : stade I et II, confinées au cortex entorhinal ; stade III et IV intéressant, de plus, l’hippocampe ; stade V et VI s’étendant à l’isocortex. Ces stades sont aujourd’hui universellement utilisés pour évaluer la gravité neuropathologique d’une maladie d’Alzheimer car ils sont liés aux stades cliniques de la maladie [37] :
les troubles mnésiques caractérisent le stade hippocampique, la démence n’apparaît qu’aux stades isocorticaux.
Chronologie des lésions
Les mutations de l’APP ou des présénilines modifient la sécrétion de peptide Aβ :
elles augmentent, en effet, le rapport Aβ 42/Aβ 40. Aucune mutation de la protéine tau, en revanche, ne provoque de maladie d’Alzheimer. Ces observations ont conduit à formuler l’hypothèse selon laquelle la perturbation du métabolisme du peptide Aβ était le primum movens, même dans les formes sporadiques de la maladie.
L’accumulation de protéine tau et la mort neuronale seraient secondaires, même si elles étaient impliquées plus directement dans l’apparition des symptômes. La suite d’interactions hypothétiques qui conduit de l’anomalie du métabolisme du peptide Aβ à la mort neuronale, a été formalisée sous la dénomination d’« hypothèse de la cascade amyloïde » [38]. Si l’on s’en tient à la lettre de cette hypothèse, l’accumulation de peptide Aβ devrait être la première lésion, précédant l’accumulation de protéine tau. Ce n’est pourtant pas généralement le cas dans les formes sporadiques de la maladie : la recherche systématique de dépôts de peptide Aβ et de protéine tau en fonction de l’âge a montré que, dans une proportion significative de la population, des DNF étaient présentes en l’absence de dépôts d’Aβ, en tout cas sur les échantillons examinés [39, 40]. Ces constatations laissent envisager la possibilité de la précession des DNF sur l’accumulation de peptide Aβ dans les formes sporadiques, une séquence qui a conduit à l’élaboration d’un modèle à deux variables : la première « le vieillissement cérébral », explique l’apparition des DNF ; la seconde, plus spécifiquement en rapport avec la maladie d’Alzheimer, serait constituée par l’accumulation de peptide Aβ, liée, par un mécanisme qui reste à préciser, à l’extension de la pathologie neurofibrillaire [40]. Les DNF, une fois formées, persistent longtemps, peut-être des décennies, d’abord dans le neurone, puis, celui-ci une fois mort, dans le milieu extracellulaire sous la forme de DNF ‘‘ fantômes ’’. Non résorbées par l’organisme, elles peuvent être suivies dans les différentes aires corticales où elles apparaissent successivement : d’abord le cortex transentorhinal et entorhinal, puis l’hippocampe (secteurs pyramidaux, avec respect au moins relatif du gyrus denté et du présubiculum). Les aires associatives sont ensuite touchées ; les aires primaires sont les dernières intéressées [24 , 36 , 41]. Des informations plus récentes laissent supposer que l’aire 19 (cortex visuel associatif) pourrait être affectée dès les premiers stades de la maladie [42]. On dispose de données bien moins précises sur la chronologie des lésions dans les noyaux sous-corticaux à l’exception
du noyau basal de Meynert [43], où des altérations neurofibrillaires sont présentes dès les premiers stades. Le dépôt diffus de peptide Aβ est être précédé ou accompagné de l’accumulation intracellulaire de peptide Aβ, une lésion bien analysée chez la souris transgénique [44].
Malgré le nombre considérable d’études neuropathologiques concernant la maladie d’Alzheimer, des chiffres incontestables de prévalence des lésions sont remarquablement peu nombreux : dans l’étude de Braak [39], critiquable du point de vue épidémiologique, des DNF étaient observées dans la moitié de la population dès l’âge de quarante-sept ans [45]. La prévalence ne cessait de croître ensuite pour atteindre 100 % chez les centenaires [46]. Celle des dépôts de peptide Aβ suivait une courbe voisine décalée de plusieurs décennies [45]. Ils étaient également constants chez les centenaires [47]. Nous manquons de données sur l’évolution temporelle des lésions. Apparaissent-elles en quelques mois au cours d’une période critique ?
Sont-elles au contraire le résultat d’une progression lente mais inexorable. Peuventelles rester stables pendant des années, des décennies ? Régressent-elles parfois ? Les récents progrès de la neuroimagerie laissent espérer la réponse à ces questions dans un avenir proche.
Les perspectives thérapeutiques telles qu’elles peuvent être suggérées par la neuropathologie
La thérapeutique peut viser les symptômes , les mécanismes ou la cause de la maladie.
Dans le premier cas, on tente de pallier les conséquences de la mort neuronale : c’est ainsi que la perte des neurones du noyau basal de Meynert provoque un déficit cholinergique que les traitements anticholinestérasiques actuels de la maladie d’Alzheimer cherchent à éviter. Différents types de greffe ont été envisagés et testés chez l’animal pour remédier ou prévenir la mort neuronale — mais il s’agit d’une méthode qui n’a pas été largement acceptée.
La compréhension, au moins partielle, des mécanismes lésionnels a ouvert une nouvelle voie thérapeutique visant à entraver les mécanismes biologiques responsables des dépôts de peptide Aβ ou de l’accumulation intracellulaire de protéine tau anormalement phosphorylée. L’hypothèse de la cascade amyloïde qui postule que la pathologie de la protéine tau est secondaire à l’accumulation du peptide Aβ est aujourd’hui presque universellement acceptée et a justifié toutes les tentatives visant à réduire sa production ou à augmenter sa clearance. Les inhibiteurs enzymatiques de la γ-secrétase sont difficiles à manier : la γ-secrétase intervient en effet dans le métabolisme de plusieurs protéines transmembranaires dont Notch, qui joue un rôle important dans les interactions cellulaires. Son inhibition pourrait donc avoir des effets secondaires importants. Des inhibiteurs de l’activité β-secretase (due à BACE 1 — beta amyloid cleaving enzyme) sont en développement et ont donné des résultats prometteurs chez les souris APP.
Des facteurs accroissant la clearance du peptide ont aussi été découverts. Les techniques immunologiques ont été les plus employées. Schenk et al. ont immunisé
l’une de ces lignées de souris (PDAPP) en leur injectant du peptide Aβ 42 synthétique par voie systémique. L’immunisation de souris jeunes prévint l’apparition des dépôts. L’immunisation tardive conduisit à l’élimination significative des dépôts déjà formés, du fait d’une activation microgliale efficace [20]. La régression de la pathologie était associée à une amélioration clinique [48]. L’étude de phase 1 du AN1792 (une combinaison d’Aβ 42 et d’adjuvant QS-21) mit en évidence des réactions secondaires — dont une méningo-encéphalite qui ne fut diagnostiquée post-mortem qu’après le début de la phase 2a, impliquant trois cents patients.
Plusieurs autres cas d’encéphalite furent identifiés et conduisirent à interrompre l’étude, comme le détaille l’article de Dubois et al (ce numéro). L’examen postmortem de patients vaccinés conclut à la rareté relative des dépôts de peptide Aβ (suggérant une régression des dépôts chez les patients, comme cela avait été observé dans le modèle murin) et à la persistance, au contraire, de l’angiopathie amyloïde et des altérations neurofibrillaires [49]. D’autres méthodes immunologiques sont aujourd’hui testées : immunoglobulines intraveineuses [50], immunisation passive [51], vaccination orale par un vecteur viral contenant l’ADN du peptide [52], utilisation d’un épitope C-terminal (qui favorise la séquestration du peptide Aβ dans le compartiment vasculaire) [53] ou au contraire d’un épitope N-terminal (qui ne stimule pas la réponse lymphocytaire T) [54]. Cependant, la perspective de prévenir ou de diminuer la pathologie de la protéine tau n’a pas été complètement abandonnée. Le lithium, par exemple, est un inhibiteur de la glycogène synthase kinase 3 (GSK 3) qui phosphoryle tau et a donné des résultats controversés chez la souris, qui n’ont pas empêché une tentative thérapeutique chez l’homme [55].
La cause de la maladie d’Alzheimer ne doit évidemment pas être confondue avec les lésions que l’on y observe. Elle n’est connue que dans le petit nombre de cas où une mutation de l’APP ou des présénilines a été mise en évidence. Ce sont ces cas que les souris transgéniques APP reproduisent. Dans les formes sporadiques, de très loin les plus fréquentes, la cause qui enclenche les mécanismes lésionnels demeure inconnue et donc inaccessible à toute tentative thérapeutique, préventive ou curative. Le modèle génétique est aveuglant et conduit à penser que, dans tous les cas, la maladie est provoquée par l’excès de production de peptide Aβ42. Dans les formes sporadiques, cette production excessive n’a jamais été prouvée et il pourrait aussi bien s’agir d’un défaut de clearance [56].
CONCLUSION
La maladie d’Alzheimer a deux composantes : l’accumulation de peptide Aβ et celle de protéines tau anormalement phosphorylée. Des lignées de souris transgéniques, exprimant pour certaines l’APP, pour d’autres la protéine tau, l’une et l’autre sous une forme mutée, ont démontré la relative indépendance des deux processus. La pathologie humaine témoigne pourtant de leur interaction par un mécanisme qui reste encore inconnu. Des résultats spectaculaires ont été obtenus chez la souris transgénique par la « vaccination » c’est-à-dire l’injection périphérique d’épitope
du peptide Aβ. Cette immunisation a conduit à la résorption ou à la prévention des dépôts. Ces résultats pourront-ils un jour s’appliquer à l’homme ? En d’autres termes, la pathologie tau est-elle totalement dépendante de l’anomalie du métabolisme du peptide Aβ ? Peut-on prévenir la maladie en prévenant les dépôts ? Les études en cours devraient rapidement apporter la réponse à cette question.
BIBLIOGRAPHIE [1] HAUW J.-J., SEILHEAN D., PIETTE F., UCHIHARA T., DUYCKAERTS C. — Les lésions de la maladie d’Alzheimer : de la morphologie à la biologie cellulaire . Bull. Acad. Natl. Méd. , 1996, 180 , 1687-1701.
[2] DUYCKAERTS C., DICKSON D. — Neuropathology of Alzheimer’s disease, in Neurodegeneration : the molecular pathology of dementia and movement disorders, Sous la direction de Dickson D. ISN Neuropath. Press , Basel, p. 47-65, 2003.
[3] DELAÈRE P., DUYCKAERTS C., HE Y., PIETTE F., HAUW J.-J. — Subtypes and differential laminar distributions of βA4 deposits in Alzheimer’s disease : relationship with the intellectual status of 26 cases. Acta Neuropathol., (Berl) , 1991, 81 , 328-335.
[4] DELAÈRE P., DUYCKAERTS C., MASTERS C., PIETTE F., HAUW J.-J. — Large amounts of neocortical βA4 deposits without Alzheimer changes in a nondemented case . Neurosci. Lett. , 1990, 116 , 87-93.
[5] JOACHIM C.L., MORRIS J.H., SELKOE D.J. — Clinically diagnosed Alzheimer’s disease : autopsy results in 150 cases . Ann. Neurol. , 1988, 24 , 50-6.
[6] ATTEMS J., JELLINGER K.A. — Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology-a pilot study . Acta Neuropathol., (Berl) , 2004, 107 , 83-90.
[7] LEVY E., CARMAN M.D., FERNANDEZ-MADRID I.J., POWER M.D., LIEBERBURG I., VAN DUINEN S.G. et al. — Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type . Science , 1990, 248 , 1124-6.
[8] BLANCHARD V., MOUSSAOUI S., CZECH C., TOUCHET N., BONICI B., PLANCHE M. et al . — Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp. Neurol. , 2003, 184 , 247-263.
[9] CASAS C., SERGEANT N., ITIER J.M., BLANCHARD V., WIRTHS O., VAN DER KOLK N. et al. —
Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model . Am. J. Pathol. , 2004, 165 , 1289-300.
[10] KIDD M. — Paired helical filaments in electron microscopy in Alzheimer’s disease . Nature , 1963, 197 , 262-268.
[11] BRION J.P., PASSAREIRO H., NUNEZ J., FLAMENT-DURAND J. — Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer. Arch. Biol. (Brux) , 1985, 95 , 229-235.
[12] GOEDERT M., SPILLANTINI M.G., POTIER M.C., ULRICH J., CROWTHER R.A. — Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats : differential expression of tau protein mRNAs in human brain . Embo. J. , 1989, 8 , 393-9.
[13] DELACOURTE A., BUEE L. — Tau pathology : a marker of neurodegenerative disorders.
Curr.
Opin. Neurol. , 2000, 13 , 371-376.
[14] DUYCKAERTS C., KAWASAKI H., DELAÈRE P., RAINSARD C., HAUW J.-J. — Fiber disorganization in the neocortex of patients with senile dementia of the Alzheimer type . Neuropath. Appl.
Neurobiol. , 1989, 15 , 233-247.
[15] BRAAK H., BRAAK E. — Neuropil threads occur in dendrites of tangle-bearing nerve cells .
Neuropathol. Appl. Neurobiol. , 1988, 14 , 39-44.
[16] HE Y., DELAERE P., DUYCKAERTS C., WASOWICZ M., PIETTE F., HAUW J.J. — Two distinct ubiquitin immunoreactive senile plaques in Alzheimer’s disease : relationship with the intellectual status in 29 cases . Acta Neuropathol. , 1993, 86 , 109-16.
[17] ARENDS Y.M., DUYCKAERTS C., ROZEMULLER J.M., EIKELENBOOM P., HAUW J.-J. — Microglia, amyloid and dementia in Alzheimer disease. A correlative study. Neurobiol. Aging , 2000, 21 , 39-47.
[18] MCGEER P.L., AKIYAMA H., ITAGAKI S., MCGEER E.G. — Complement activation in amyloid plaques in Alzheimer’s dementia. Neurosci. Letters , 1989, 107 , 341-346.
[19] DICKSON D.W., LEE S.C., MATTIACE L.A., YEN S.C., BROSNAN C. — Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease . Glia. , 1993, 7 , 75-83.Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T. et al. — Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse.
Nature , 1999, 400 , 173-177.
[20] SCHENK D., BARBOUR R., DUNN W., GORDON G., GRAJEDA H., GUIDO T., et al. — Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse.
Nature , 1999, 400 , 173-177.
[21] UCHIHARA T., DUYCKAERTS C., HE Y., KOBAYASHI K., SEILHEAN D., AMOUYEL P. et al . —
ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain.
Neurosci. Lett. , 1995, 195 , 5-8.
[22] DUYCKAERTS C., HAUW J.-J., BASTENAIRE F., PIETTE F., POULAIN C., RAINSARD V. et al. —
Laminar distribution of neocortical plaques in senile dementia of the Alzheimer type . Acta Neuropathol (Berl) , 1986, 70 , 249-256.
[23] DELATOUR B., BLANCHARD V., PRADIER L., DUYCKAERTS C. — The innervation of senile plaques : a link between amyloid and neurofibrillary pathology ? in Alzheimer’s disease and related disorders. Annual 2004. Sous la direction de Gauthier S., Scheltens P., Cummings J. L.
Martin Dunitz, London. p. 1-19, 2003.
[24] METSAARS W.P., HAUW J.J., WELSEM M.E., DUYCKAERTS C. — A grading system of Alzheimer disease lesions in neocortical areas. Neurobiol. Aging , 2003, 24 , 563-572.
[25] GOMEZ-ISLA T., HOLLISTER R., WEST H., MUI S., GROWDON J.H., PETERSEN R.C. et al. —
Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease . Ann.
Neurol. , 1997, 41 , 17-24.
[26] GRIGNON Y., DUYCKAERTS C., BENNECIB M., HAUW J.-J. — Cytoarchitectonic alterations in the supramarginal gyrus of late onset Alzheimer’s disease . Acta Neuropathol., (Berl) , 1998, 95 , 395-406.
[27] TERRY R.D., PECK A., DETERESA R., SCHECHTER R. — Some morphometric aspects of the brain in senile dementia of the Alzheimer type . Ann. Neurol. , 1981, 10 , 184-192.
[28] DUYCKAERTS C., LLAMAS E., DELAÈRE P., MIELE P., HAUW J.-J. — Neuronal loss and neuronal atrophy. Computer simulation in connection with Alzheimer’s disease. Brain Res. , 1989, 504 , 94-100.
[29] REGEUR L., BADSBERG JENSEN G., PAKKENBERG H., EVANS S.M., PAKKENBERG B. — No global neocortical nerve cell loss in brains from patients with senile dementia of Alzheimer’s type.
Neurobiol. Aging , 1994, 15 , 347-352.
[30] TERRY D., MASLIAH E., HANSEN L.A. — Structural basis of the cognitive alterations in Alzheimer disease, in Alzheimer disease, Sous la direction de Terry R. D., Katzman R., Bick K.L., Raven Press , New York. p. 179-196, 1994.
[31] SCHEFF S.W., SPARKS L., PRICE D.A. — Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer’s disease . Ann. Neurol. , 1993, 34 , 356-61.
[32] MASLIAH E., TERRY R.D., DETERESA R.M., HANSEN L.A. — Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci. Lett. , 1989, 103 , 234-239.
[33] BONCRISTIANO S., CALHOUN M.E., HOWARD V., BONDOLFI L., KAESER S.A., WIEDERHOLD K.H. et al. — Neocortical synaptic bouton number is maintained despite robust amyloid deposition in APP23 transgenic mice . Neurobiol. Aging , 2005, 26 , 607-13.
[34] DICKSON D.W., CRYSTAL H.A., BEVONA C., HONER W., VINCENT I., DAVIES P. — Correlations of synaptic and pathological markers with cognition of the elderly . Neurobiol. Aging , 1995, 16 , 285-304.
[35] THAL D.R., RUB U., ORANTES M., BRAAK H. — Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology , 2002, 58 , 1791-1800.
[36] BRAAK H., BRAAK E. — Neuropathological stageing of Alzheimer-related changes . Acta
Neuropathol. (Berl) , 1991, 82 , 239-259.
[37] BRAAK H., DUYCKAERTS C., BRAAK E., PIETTE F. — Neuropathological staging of Alzheimerrelated changes correlates with psychometrically assessed intellectual status, in Alzheimer’s disease : advances in clinical and basic research , Sous la direction de Corain B., Iqbal K.,
Nicolini M., Winblad B., Wisniewski H., Zatta P., John Wiley & Sons, Chichester. p. 131-137, 1993.
[38] HARDY J. — An ‘anatomical cascade hypothesis’ for Alzheimer’s disease. Trends in Neuros- ciences , 1992, 15 , 200-201.
[39] BRAAK H., BRAAK E. — Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging , 1997, 18 , 351-357.
[40] DUYCKAERTS C., HAUW J.-J. — Prevalence, incidence and duration of Braak’s stages in the general population : can we know ? Neurobiol. Aging , 1997, 18 , 362-369.
[41] DELACOURTE A., DAVID J.P., SERGEANT N., BUEE L., WATTEZ A., VERMERSCH P. et al. — The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease.
Neurology , 1999, 52 , 1158-1165.
[42] MCKEE A.C., AU R., CABRAL H.J., KOWALL N.W., SESHADRI S., KUBILUS C.A. et al. — Visual association pathology in preclinical Alzheimer disease . J. Neuropathol. Exp. Neurol. , 2006, 65 , 621-30.
[43] SASSIN I., SCHULTZ C., THAL D.R., RUB U., ARAI K., BRAAK E. et al. — Evolution of
Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert . Acta Neuropathol. (Berl) , 2000, 100 , 259-69.
[44] LANGUI D., GIRARDOT N., EL HACHIMI H., ALLINQUANT B., BLANCHARD V., PRADIER L. et al.
— Subcellular topography of neuronal A-beta peptide in APPxPS1 transgenic mice . Am. J.
Pathol. , 2004, 165 , 1465-1477.
[45] DUYCKAERTS C., HAUW J.-J. — Prevalence, incidence and duration of Braak’s stages in the general population : can we know ? Neurobiol. Aging , 1997, 18 , 362-9 discussion 389-92.
[46] HAUW J.-J., VIGNOLO P., DUYCKAERTS C., BECK M., FORETTE F., HENRY J.F. et al. — Étude neuropathologique de 12 centenaires : la fréquence de la démence sénile de type Alzheimer n’est pas particulièrement élevée dans ce groupe de personnes très agées. Rev. Neurol. (Paris) , 1986, 142 , 107-115.
[47] DELAERE P., HE Y., FAYET G., DUYCKAERTS C., HAUW J.J. — Beta A4 deposits are constant in the brain of the oldest old : an immunocytochemical study of 20 French centenarians .
Neurobiol. Aging , 1993, 14 , 191-4.
[48] JANUS C., PEARSON J., MCLAURIN J., MATHEWS P.M., JIANG Y., SCHMIDT S.D. et al. — A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease . Nature , 2000, 408 , 979-82.
[49] NICOLL J.A., BARTON E., BOCHE D., NEAL J.W., FERRER I., THOMPSON P. et al. — Abeta species removal after abeta42 immunization . J Neuropathol Exp Neurol , 2006, 65 , 1040-8.
[50] SOLOMON B. — Intravenous immunoglobulin and Alzheimer’s disease immunotherapy . Curr.
Opin. Mol. Ther. , 2007, 9 , 79-85.
[51] GARDBERG A.S., DICE L.T., OU S., RICH R.L., HELMBRECHT E., KO J. et al. — Molecular basis for passive immunotherapy of Alzheimer’s disease . Proc. Natl. Acad. Sci. U S A , 2007, 104 , 15659-64.
[52] MOURI A., NODA Y., HARA H., MIZOGUCHI H., TABIRA T., NABESHIMA T. — Oral vaccination with a viral vector containing Abeta cDNA attenuates age-related Abeta accumulation and memory deficits without causing inflammation in a mouse Alzheimer model . Faseb J. , 2007, 21 , 2135-48.
[53] GRAY A.J., SAKAGUCHI G., SHIRATORI C., BECKER A.G., LAFRANÇOIS J., AISEN P.S. , et al. —
Antibody against C-terminal Abeta selectively elevates plasma Abeta . Neuroreport , 2007, 18 , 293-6.
[54] MORETTO N., BOLCHI A., RIVETTI C., IMBIMBO B.P., VILLETTI G., PIETRINI V. et al. —
Conformation-sensitive antibodies against alzheimer amyloid-beta by immunization with a thioredoxin-constrained B-cell epitope peptide . J. Biol. Chem. , 2007, 282 , 11436-45.
[55] ZHU X., AVILA J., PERRY G., SMITH M.A. — Treating the lesions, not the disease . Am. J.
Pathol. , 2007, 170 , 1457-9.
[56] FARRIS W., SCHUTZ S.G., CIRRITO J.R., SHANKAR G.M., SUN X., GEORGE A. et al. — Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. J. Neurosci. , 2007, 27 , 2866-2875.
DISCUSSION
M. Bernard PESSAC
Quelle(s) est(sont) la(es) relation(s) entre le peptide neurotoxique et le type neuronal affecté ?
Le rôle directement toxique du peptide Aβ sur le neurone est discuté. D’après l’hypothèse de la cascade amyloïde, c’est l’accumulation de peptide Aβ qui provoque la mort neuronale à la suite d’une série de réactions. Cette hypothèse peut sembler confortée par les données obtenues à partir de cultures cellulaires : in vitro , en effet, le peptide Aβ, probablement oligomérisé, ou certains de ses fragments, provoque la mort cellulaire.
Chez l’homme, les dépôts de peptide Aβ sont parfois abondants dans des régions où aucun signe de mort neuronale n’est visible. La dégénérescence neurofibrillaire, c’est-à- dire l’agrégation intracellulaire de protéine tau anormalement phosphorylée, constituet-elle, dans ces conditions, une étape de la neurotoxicité ? Le rapport entre l’accumulation de peptide Aβ et la dégénérescence neurofibrillaire reste encore aujourd’hui mal compris.
La simple surproduction d’APP humaine mutée chez la souris transgénique ne suffit pas à déclencher la pathologie neurofibrillaire. On peut aussi rappeler que les données actuelles laissent penser que les dégénérescences neurofibrillaires précèdent chez
l’homme les dépôts de peptide Aβ. Peut-on définir un type neuronal sélectivement affecté ? Les cellules pyramidales de taille moyenne de l’hippocampe ou de la couche III de l’isocortex apparaissent particulièrement vulnérables ; elles sont glutamatergiques.
Mais les neurones du noyau basal de Meynert, multipolaires et cholinergiques, sont aussi lésés comme le sont les cellules pigmentées noradrénergiques du locus coeruleus. Il n’y a donc pas de population neuronale spécifiquement vulnérable.
M. Bernard LECHEVALIER
Quel est le rapport entre l’artériopathie (angiopathie) amyloïde cérébrale qui se manifeste par des hémorragies cérébrales à répétition chez les personnes âgées et la maladie d’Alzheimer, plus spécialement l’angiopathie dyshorique, observée microscopiquement dans les régions postérieures du cerveau. S’agit-il de la même substance amyloïde A β 42 ?
L’analyse du peptide Aβ dans les plaques séniles et dans la paroi des vaisseaux, et la production de nouveaux modèles transgéniques ont permis d’élucider, au moins en partie, les rapports entre l’angiopathie amyloïde et la maladie d’Alzheimer. L’une et l’autre sont souvent associées puisque la présence de peptide Aβ dans les parois vasculaires est avérée dans la très grande majorité des cas de maladie d’Alzheimer. Cependant, chez certains patients, c’est la pathologie vasculaire qui est prédominante ; la multiplication des hémorragies ou des infarctus de petite taille est la cause directe de la démence.
C’est le cas, par exemple, au cours de la mutation Hollandaise (Glu693Gln). À la différence de ce qui est observé dans les plaques séniles (où le peptide Aβ long de 42 acides aminés constitue le précipité initial), la paroi des vaisseaux s’enrichit en peptide Aβ40.
Chez les souris transgéniques qui surexpriment l’APP humain affecté par la mutation hollandaise Glu693Gln, la sécrétion du peptide Aβ40 est particulièrement élevée [1]. Ce peptide, plus soluble que le peptide Aβ42, précipite plus lentement dans le parenchyme et peut donc être drainé vers les vaisseaux dans la paroi desquels il s’accumule — reproduisant le phénotype de l’angiopathie amyloïde. Des plaques séniles parenchymateuses, analogues à celles qui sont observées dans la maladie d’Alzheimer peuvent être observées si, outre le premier gène muté — APP Glu693Gln —, le gène humain muté de la préséniline est cotransfecté. Ce nouveau transgène augmente la quantité de peptide Aβ42 produit. Celui-ci précipite dans le parenchyme avant d’atteindre les vaisseaux ce qui explique que les plaques parenchymateuses soient abondantes dans ce nouveau modèle.
Il apparaît donc que c’est la balance Aβ42/Aβ40 qui détermine au moins en partie le tropisme du peptide Aβ pour le parenchyme (concentration élevée d’Aβ42 peu soluble) ou pour les vaisseaux (concentration élevée d’Aβ40 plus soluble).
M. Jacques CAEN
Existe-t-il des modéles animaux autres que les souris transgéniques pour apprécier comment les différents neurones résistent ou pas à la toxicité du peptide de 42 acides aminés.
L’environnement neuronal a-t-il un rôle à jouer ? Comment s’opère l’addition de deux acides aminés (quels sont-ils ?) au peptide de 40 acides aminés transmembranaires ?
De nombreux modèles animaux ont été proposés avant l’avènement des souris transgé- niques : toxiques ou lésionnels, par exemple, à l’époque où l’hypothèse cholinergique faisait jouer un rôle central à la destruction du noyau basal de Meynert qui assure
l’innervation cholinergique de l’isocortex ; spontanés surtout, car des dépôts de peptide Aβ ont été observés dans de nombreuses espèces de mammifères, du vieux chien au vieil ours. En France, le lémurien, un primate prosimien, a été particulièrement étudié.
L’avantage du modèle spontané — l’absence de surexpression artificielle d’un protéine humaine mutée — est contrebalancé par ses inconvénients : approche principalement descriptive, nécessitant un temps d’élevage prolongé, les altérations n’apparaissant que chez les animaux âgés. Aucune étude systématique n’a, à ma connaissance, tenté de déterminer si certaines espèces étaient totalement dépourvues de dépôt de peptide Aβ.
L’environnement neuronal joue-t-il un rôle dans la pathogénie ? Sans aucun doute :
l’astrocyte entoure la plaque de nombreux prolongements ; la cellule microgliale est au contact de la substance amyloïde qu’elle phagocyte (ou selon une autre hypothèse qu’elle produit à partir du dépôt de peptide Aβ). Il est cependant bien établi aujourd’hui que la production neuronale de peptide Aβ suffit à reproduire les lésions amyloïdes. Il ne semble pas que la neuroglie sécrète le peptide. Le clivage de l’extrémité C-terminale de l’APP par le complexe gamma-secrétase produit le peptide Aβ42 ou 40 — le premier se termine par les acides aminés isoleucine et alanine qui suivent les deux valines qui terminent le peptide Aβ40. La raison pour laquelle ce clivage peut se produire à deux endroits différents et la régulation de cette coupure différentielle ne sont pas connues. La possibilité d’un ajout de deux acides aminés supplémentaires au peptide Aβ40 qui serait initialement produit n’est pas retenue aujourd’hui.
M. René MORNEX
Cette excellente analyse physiopathologique ouvre-t-elle des voies d’explication de l’augmentation de la prévalence dans ces trente dernières années ?
La prévalence de la maladie a sans aucun doute augmenté avec le vieillissement de la population. Il est généralement admis que ce n’est pas le cas de son incidence, à âge égal.
Cette question permet de signaler que la maladie d’Alzheimer a un passé prestigieux.
Parmi ses victimes célèbres, citons Emmanuel Kant, Joseph Haydn, ou Lucullus, général Romain fameux dont la biographie figure dans les Vies Parallèles de Plutarque : ‘‘ Lucullus eut l’entendement perdu et bouché, de sorte que, de son vivant même, son frère dut administrer sa fortune ’’.
M. Jean-Pierre NICOLAS
L’élimination des produits de dégradation de l’APP (en particulier de l’A β 42 trouvé dans les plaques séniles) peut être réalisée par trois voies : enzymatique, vasculaire et par le liquide céphalorachidien. Avec l’âge, la vitesse de renouvellement du LCR diminue de façon importante. Pensez-vous que cela puisse jouer un rôle dans la genèse de la maladie ?
La production du peptide Aβ a sans doute fait l’objet d’un nombre plus grand d’études que son élimination, qui pourrait pourtant être un facteur étiologique important. On admet aujourd’hui, à la suite des travaux de Roy Weller et de son équipe, que le peptide est drainé dans les espaces de Virchow-Robin qui communique avec les espaces sousarachnoïdiens. Au cours de la maladie d’Alzheimer, la concentration du peptide Aβ42 dans le liquide céphalo-rachidien (LCR) diminue, contrairement à ce que l’on pourrait attendre. Cette baisse est généralement attribuée à l’effet des dépôts constitués qui capteraient le peptide Aβ42 secrété par les neurones. L’hypothèse d’une diminution de la
clearance provoquée par une réduction du débit du LCR est très intéressante et n’a, à ma connaissance, jamais été soulevée ; elle mériterait d’être explorée.
M. François-Bernard MICHEL
A-t-il été observé, au stade initial des lésions locales, un processus inflammatoire (activation de cytokines) ?
Cette question permet de considérer un chapitre important de la pathogénie de la maladie d’Alzheimer et d’envisager les tentatives thérapeutiques qu’elle a suscitées. La cellule microgliale (terme qu’on peut utiliser ici comme synonyme de macrophage) qui accompagne toujours le dépôt amyloïde du cœur de la plaque sénile secrète des cytokines et provoque l’apparition d’une réaction inflammatoire humorale en l’absence de lymphocytes. Cette inflammation à bas bruit, focalisée à la plaque sénile, a été considérée comme potentiellement toxique et a justifié l’utilisation de thérapeutiques anti-inflammatoires qui n’ont jamais fait la preuve de leur utilité. Le rôle de la cellule microgliale a été débattu : elle a été naguère jugée responsable de la transformation amyloïde du peptide Aβ ; on a pu penser, au contraire, qu’elle résorbait les fibrilles d’amyloïde. L’hypothèse de Dale Schenk, qu’on puisse stimuler les macrophages pour résorber les plaques séniles en provoquant une immunisation contre le peptide Aβ (la ‘‘ vaccination ’’), s’est révélée exacte au moins chez la souris transgénique et laisse penser qu’une partie des réactions inflammatoires soit bénéfique.
* Laboratoire de neuropathologie Raymond Escourolle, Groupe hospitalier Pitié-Salpêtrière, 47, boulevard de l’Hôpital, 75651 Paris Cedex 13, e-mail : charles.duyckaerts@psl.aphp.fr Tirés-à-part : Professeur Charles DUYCKAERTS, même adresse. Article reçu et accepté le 11 février 2008.
Bull. Acad. Natle Méd., 2008, 192, no 2, 303-321, séance du 12 février 2008