
Résumé
L’étude des tumeurs par la technique de « biopuces » ou « DNA-array » permet d’obtenir le profil instantané du niveau d’expression de plusieurs centaines de gènes. Afin de tester l’intérêt de cette technique dans le diagnostic des lymphomes, nous avons analysé 17 biopsies ganglionnaires à l’aide d’une membrane de type « macro-array » contenant 217 gènes impliqués dans l’apoptose ou le cycle cellulaire. Huit échantillons correspondaient à des cellules lymphoïdes triées par billes magnétiques : cellules B de 2 lymphomes folliculaires (LF) et de 2 lymphomes B diffus à grandes cellules (LDGC), cellules B et cellules T de 2 adénites, cellules B vierges de phénotype CD19+/CD38-/IgD+ et cellules B des centres germinatifs de phénotype CD19+/CD38+/IgD- de 2 amygdales bénignes. Neuf échantillons étaient des fragments tissulaires entiers correspondant histologiquement à 4 LF, 2 LDGC et 3 adénites. Dans le groupe des cellules triées, les profils d’expression étaient statistiquement corrélés à la nature des échantillons, permettant de séparer les LF, les LBGC, et les échantillons bénins. Les corrélations étaient moins nettes dans le groupe des tissus. Pour vérifier la fiabilité de la technique de biopuce, nous avons analysé en parallèle par immunohistochimie (IHC) l’expression des caspases -2, -3, -8, -7 et -9. Les résultats des deux méthodes montraient une bonne corrélation. Ces résultats suggèrent une application possible des biopuces pour le diagnostic des lymphomes malins.
Summary
Microarray technology has recently led to the identification of molecular prognostic subgroups in non Hodgkin’s lymphomas. In order to determine the usefulness of ready-made macroarrays as routine diagnosis tools in haemato-pathology, we have analysed lymph node biopsies using a cDNA macroarray containing genes involved in apoptosis, including caspases. Nine biopsy specimens were analysed on total frozen tissues : 4 samples of B-cell follicular lymphoma (FL), two of B-cell diffuse large cell lymphoma (DLCL), and three of non-neoplastic lymph nodes from benign lymphadenitis. Eight cell populations were sorted from fresh tissues : malignant B-cells from 2 FL cases and 2 DLCL cases, reactive B-cells from 1 benign lymph nodes, reactive T-cells from 1 benign lymph node, virgin (mantle zone) B-cells and germinal center (GC) B-cells from benign tonsils. Immunohistochemistry (IHC) on paraffin sections was performed for localization of caspases 2, 3, 4, 7, 8, and 9. In the clustered array data, sorted cells from samples sharing common histological lesions grouped together, whereas the array/histology correlation was less satisfactory for tissues. The expression profiles of both array and IHC methods were correlated for most caspases and samples. Variations in array profiles of sorted cell populations can be statistically associated with specific histological features, suggesting a possible diagnostic application of ready-made « Apoptosis macroarrays » in haematopathology.
INTRODUCTION
L’identification exhaustive des nombreux gènes dont l’expression est altérée dans les cellules cancéreuses et qui jouent probablement un rôle dans la croissance tumorale représente un objectif majeur en cancérologie. La recherche efficace d’altérations significatives de l’expression génique des cellules cancéreuses impose d’analyser simultanément plusieurs centaines ou milliers de gènes. Dans cette optique a été développée récemment une nouvelle technique dite « biopuce » ou « DNA-array » ou « membrane à haute densité » [1]. Il s’agit d’un support de nylon sur lequel est fixée une multitude d’échantillons calibrés d’acide désoxyribonucléique complé- mentaire (ADNc), spécifiques de différents gènes. Ce filtre est hybridé avec une sonde dite « complexe » dérivée de l’acide ribonucléique messager (ARNm) extrait des cellules ou tissus à tester. Les signaux d’hybridation donnent alors le profil instantané global de l’expression génique dans le tissu dont la sonde est dérivée.
Cette technique permet l’analyse simultanée, quantitative et comparative, de l’expression d’un nombre de gènes considérable [2, 3], et de comparer les profils d’expression dans différents types cellulaires ou tissus [4, 5].
Parmi les gènes pouvant bénéficier de cette approche, les gènes régulateurs de la mort cellulaire programmée ou apoptose apparaissent comme une cible de choix en cancérologie. L’apoptose est un mécanisme physiologique actif d’autodestruction cellulaire, qui joue un rôle important dans le développement harmonieux de tout
organisme multicellulaire. Ce processus correspond à une cascade biochimique faisant intervenir des récepteurs membranaires, des régulateurs et des effecteurs, notamment une famille d’enzymes protéolytiques appelées caspases [6].
Des anomalies d’expression de régulateurs de l’apoptose sont présentes dans divers types de cancers, et en particulier dans les lymphomes malins non-hodgkiniens (LMNH), groupe de tumeurs d’une grande diversité clinico-pathologique et molé- culaire [7]. L’expression de la protéine anti-apoptotique BCL-2 est augmentée dans plus de 85 % des LMNH de type folliculaire en raison d’un réarrangement du gène BCL-2 [8]. Le rôle possible d’anomalies de l’apoptose dans la pathogénie des autres types de LMNH est suggéré par leur résistance fréquente à la stimulation du récepteur membranaire pro-apoptotique FAS [9, 10], ainsi que par une forte expression de la caspase-3 dans les cellules lymphoïdes B des centres germinatifs (CGs) et dans les LMNH dérivés des CGs [11, 12]. Cette caspase semble être un médiateur crucial de l’apoptose dans les cellules lymphomateuses [13].
Étant donnée l’importance potentielle de l’apoptose dans la physiopathologie des lymphomes malins, nous avons supposé que les niveaux d’expression des gènes régulateurs apoptotiques pouvaient être corrélés à la diversité histologique des lymphomes, et constituer donc un outil diagnostique en hématopathologie. Pour vérifier cette hypothèse, nous avons analysé des biopsies de ganglions lymphatiques par une technique de membrane à haute densité permettant l’analyse d’un ensemble de gènes impliqués dans l’apoptose.
MATÉRIELS ET MÉTHODES
Préparation d’ARN à partir d’échantillons tissulaires et cellulaires
L’ARN utilisé pour la synthèse des sondes a été purifié à partir d’échantillons biopsiques tissulaires ou de cellules triées, après vérification histologique de la nature du tissu d’origine. Les lymphomes ont été typés suivant la classification de l’OMS [7]. Les échantillons ont été divisés en 2 groupes. Ceux du premier groupe ont été analysés sur tissu total congelé : quatre cas de lymphomes B de type folliculaire (LF-1 à LF-4), deux cas de lymphomes B diffus à grandes cellules (LDGC-1 et LDGC-2), et trois cas d’adénite réactionnelle bénigne. Le deuxième groupe comportait 8 échantillons biopsiques à partir desquels des populations cellulaires B CD20+ /CD3- ont été triées par technique de billes magnétiques : 2 cas de LF (LF-1 et LF-2), 2 cas de LDGC (LDGC-1 et LDGC-2), 2 populations de cellules B totales issues de 2 ganglions d’adénite, une population de cellules B vierges de la zone du manteau (ZM) de phénotype CD19+ / IgD+ / CD38- ainsi que des cellules B du CG de phénotype CD19+ / IgD- / CD38+ issues d’amygdales bénignes. La vérification de la qualité de l’ARN a été effectuée par électrophorèse en gel dénaturant et par une réaction de transcription inverse et amplification (RT-PCR) sur un gène contrôle, la β2-microglobuline.
Hybridation des membranes à haute densité
Nous avons utilisé une membrane à haute densité de type « macro-array » TM (ATLAS Human Apoptosis Array ; Clontech Laboratories Inc, Palo Alto, CA Etats-Unis) comportant 217 échantillons d’ADNc correspondant principalement à des gènes humains impliqués dans la régulation de la mort cellulaire programmée, ainsi que dans le contrôle du cycle cellulaire. Les contrôles négatifs sont des gènes non-humains (ADN plasmidiques et de bactériophages), les contrôles positifs sont constitués par des gènes humains exprimés de façon ubiquitaire. Pour chaque échantillon d’ARN, une sonde dite « complexe » a été synthétisée à partir de 25 µg d’ARN total par transcription inverse et incorporation de phosphore radioactif de type [33P]. Les membranes ont été hybridées pendant 48 heures à 68° C.
Analyse bio-informatique et quantification des données
Les signaux de radioactivité des membranes hybridées ont été visualisés à l’aide d’un écran phosphore (FUJI BAX 1 500), et les signaux d’hybridation quantifiés avec une version modifiée du logiciel « HDG Analyser » (Bio-image). Ce logiciel détectait automatiquement les signaux, il déterminait leurs formes et leurs limites, et les quantifiait individuellement en réalisant une soustraction du bruit de fond. L’intensité de chaque signal était proportionnelle au niveau d’expression du gène concerné.
Les données étaient ensuite soumises à une normalisation, qui consistait à diviser la valeur de l’intensité de chaque signal par la somme des intensités de 7 gènes d’expression ubiquitaire présents sur la même membrane. Ceci permettait d’obtenir une valeur relative qui n’était pas affectée par d’éventuelles variations des conditions expérimentales, et donc de comparer les valeurs obtenues dans différentes expériences.
Pour l’analyse statistique, nous avons utilisé un algorithme informatique de classification hiérarchique basé sur le coefficient de corrélation de Pearson. Ce coefficient permet d’évaluer les degrés de similitude entre les profils d’expression de chaque échantillon et de représenter les résultats sous forme d’arbres statistiques ou dendrogrammes, dans lesquels les échantillons dont les profils sont les plus proches se disposent à proximité les uns des autres.
Immunohistochimie
Nous avons utilisé des anticorps monoclonaux spécifiques dirigés contre les caspases -2, -3, -7, -8, et -9. La technique a été effectuée sur des coupes en paraffine après démasquage antigénique par la chaleur.
RÉSULTATS
Contrôles
Les différences des niveaux de transcription d’un gène à l’autre étaient importantes (Fig. 1), avec des valeurs d’intensité des signaux positifs s’étendant sur 3 ou 4 ordres de grandeur en échelle logarithmique (Fig. 2). Les résultats de quantification ont été représentés sous une forme graphique (tableau à double entrée), en utilisant une échelle logarithmique de couleur (Fig. 2). Les clones qui correspondaient aux gènes non humains (contrôles négatifs) ne donnaient aucun signal, indiquant l’absence d’hybridation non spécifique. Les clones correspondant aux gènes exprimés de façon ubiquitaire (contrôles positifs) donnaient en général des niveaux d’expression élevés. La comparaison des profils d’expression entre les cellules des CGs et les cellules B vierges montraient quelques différences prévisibles, puisque les clones correspondant à certains facteurs de transcription, tels que AP-1 , Jun-B et à la protéine pro-apoptotique
BAX étaient exprimés à des taux plus élevés dans les cellules du CG que dans les cellules B vierges, ce qui est en accord avec les données de la littérature [14].
Profils d’expression génique dans les échantillons lymphoïdes
Deux arbres statistiques ont été obtenus, correspondant au groupe des cellules purifiées et au groupe des tissus, sur lesquels les échantillons ont des profils d’expression génique d’autant plus similaires qu’ils sont reliés sur le graphe par des branches courtes alors que les échantillons dont les profils sont éloignés sont reliés par des branches longues (Fig. 3).
Dans le groupe des cellules triées, les 2 échantillons de cellules B issues de ganglions bénins étaient regroupés étroitement, de même que les échantillons de LDGC. Les échantillons de LF étaient distribués dans des branches terminales longues, suggé- rant que leurs profils d’expression génique étaient plus hétérogènes d’une tumeur à l’autre. Les échantillons de LF et de cellules B normales étaient regroupés à proximité des échantillons de cellules B vierges. Les profils d’expression des échantillons de LDGC étaient très distincts de ceux des échantillons de LF mais proches des échantillons de cellules B des CGs. Globalement, les échantillons montrant des lésions histologiques communes se groupaient ensemble avec une tendance plus nette dans le groupe des cellules purifiées que dans celui des tissus (Fig. 3).
Corrélations entre les profils d’expression des caspases déterminés par biopuces et immunohistochimie
Afin de valider les données des biopuces, nous avons comparé les résultats d’hybridation des gènes codant pour les caspases et l’expression protéique des caspases en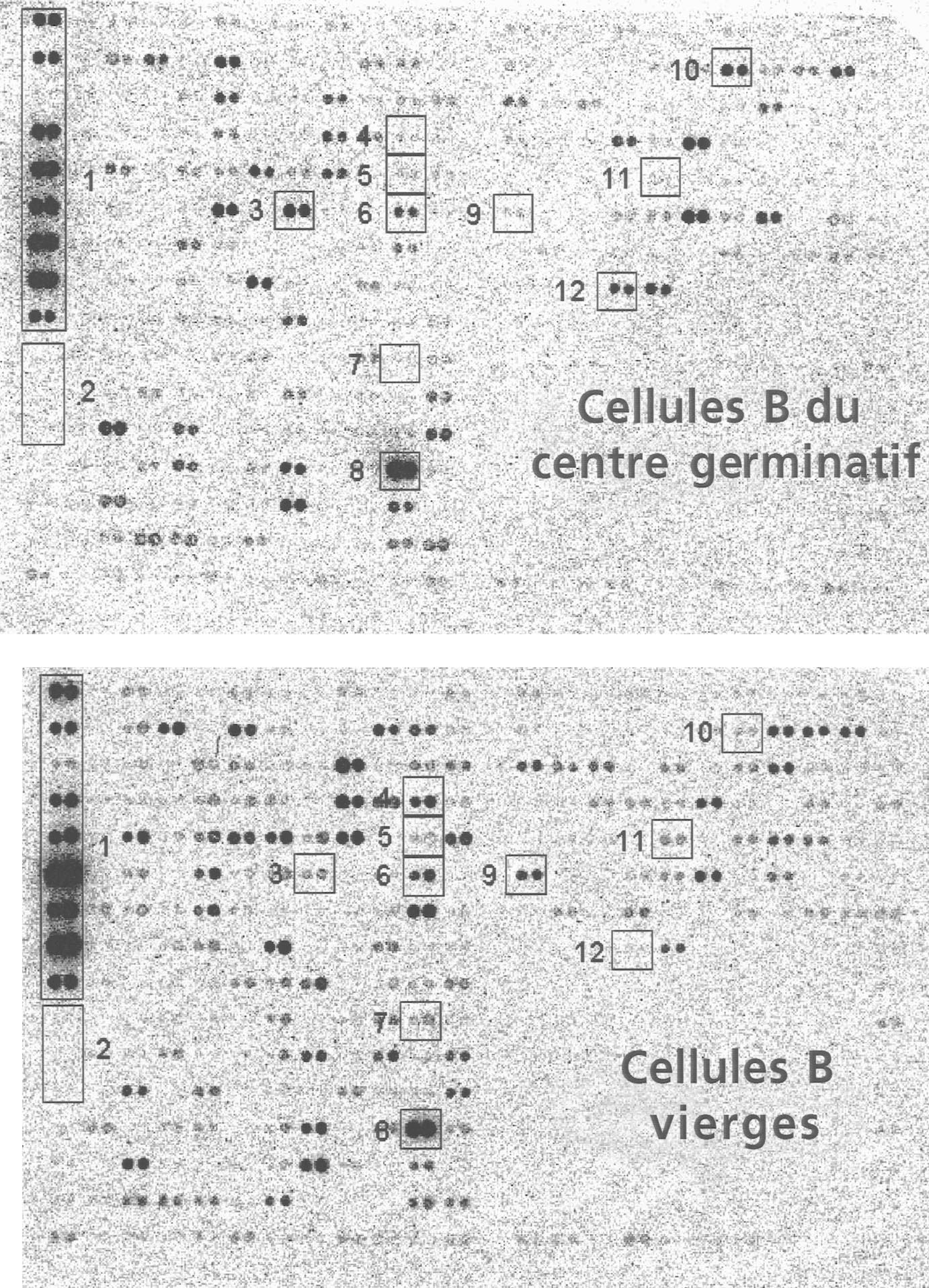
FIG. 2. — Résultats des profils d’expression génique obtenus par biopuces après analyse informatique des signaux.
La représentation en échelle de couleur permet de visualiser pour chaque gène les différences d’expression entre les échantillons.
FIG. 3. — Arbres statistiques de classification (dendrogrammes) en fonction des similitudes des profils d’expression.
Les échantillons ont un profil d’expression d’autant plus proches qu’ils sont reliés par des branches courtes.
immunohistochimie. Par les deux méthodes, nous avons obtenu des profils d’expression globalement concordants pour l’ensemble des caspases dans les différents échantillons.
L’ARN de la caspase-2 était faiblement exprimé au niveau des cellules B vierges, alors que les cellules du CG étaient négatives. Dans les échantillons tissulaires de LMNH positifs en hybridation, l’IHC montrait que la caspase-2 était en effet positive dans la plupart des cellules lymphomateuses. Elle montrait dans les follicules lymphoïdes des tissus bénins une positivité des cellules B vierges de la ZM et une négativité des cellules B du CG.
L’ARN de la caspase-3 était exprimé à des niveaux plus élevés dans les cellules du CG que dans les cellules B vierges. Le même profil était obtenu avec l’IHC. L’ARN de la caspase-7 était détecté dans 5/10 échantillons de LMNH et dans 4/7 échantillons bénins. Les cellules du CG et de la ZM étaient négatives. L’IHC confirmait la positivité des cellules lymphomateuses au niveau des échantillons montrant une expression d’ARN, et confirmait la négativité des cellules du CG et de la ZM des follicules normaux.
L’expression d’ARN de la caspase-8 dans les cellules B vierges était plus élevée que dans les cellules du CG. L’IHC confirmait ce dernier résultat. L’ARN de la caspase-9 était exprimé dans 7/10 échantillons de LMNHs et au niveau de 2/7 échantillons de tissu bénin, tandis que les cellules du CG et celles de la ZM étaient négatives. L’IHC confirmait l’expression de la caspase-9 dans les cellules tumorales de LMNH, ainsi que la négativité des cellules du CG et des cellules de la ZM.
COMMENTAIRES
La technologie des « biopuces » offre des perspectives nouvelles d’investigation en cancérologie. Dans le domaine particulier des lymphomes malins, une analyse récente utilisant une biopuce de 18 000 gènes a permis l’identification de sousgroupes pronostiques de patients en fonction des profils d’expression des tumeurs correspondantes [15]. Le but de notre travail était de vérifier si cette technologie pouvait être utilisée comme un outil de diagnostic en hématopathologie. Nous nous sommes plus particulièrement intéressés aux cellules B du CG et aux cellules B vierges de la ZM, deux populations lymphoïdes qui peuvent être analysées à la fois par biopuce et par IHC. Nos résultats des niveaux d’expression d’ARN des gènes BAX , de la caspase-3, de facteurs de transcription et des antigènes nucléaires de prolifération sont en accord avec les données de la littérature [11, 12, 14]. Il existait par ailleurs une bonne corrélation entre les profils d’expression analysés par IHC et par biopuces, ce qui suggère une bonne fiabilité de cette dernière approche. Le profil d’expression des caspases a pu être caractérisé plus précisément grâce à l’association des deux techniques.
La caspase-2 peut agir comme un inducteur ou un inhibiteur de l’apoptose en raison de l’existence de différentes formes transcriptionnelles [16, 17]. Nos résultats montrent une expression plus élevée de la caspase-2 dans les cellules B vierges que dans celles du CG, ce qui est en faveur d’une fonction prédominante de survie de la caspase-2 dans les cellules B. Les caspases effectrices telles que la caspase-3, caspase-6 et caspase-7 sont activées par d’autres caspases et se situent en aval dans la voie de mort cellulaire [18-20]. Une forte expression de la caspase-3 dans les cellules lymphoïdes normales du CG a été décrite dans la littérature [11, 12]. Bien que la caspase-6 et la caspase-7 puissent remplacer et potentialiser la caspase-3 dans certains types cellulaires et/ou dans certaines voies apoptotiques [19], la faible expression des caspases-6 et -7 que nous avons observée dans les échantillons normaux en comparaison de la caspase-3 suggère que cette dernière est la principale caspase effectrice dans ces cellules.
Les caspases initiatrices, telles que la caspase-8, -9, et -10, sont préférentiellement activées par un stimulus provenant d’un récepteur membranaire, et sont capables d’activer les caspases effectrices [21-23]. La caspase-9, impliquée dans la voie mitochondriale d’activation des caspases [23], n’a pas été détectée à des niveaux significatifs dans les cellules et tissus lymphoïdes normaux, ce qui suggère que cette
voie mitochondriale ne joue pas un rôle majeur dans le processus de mort cellulaire physiologique du système lymphoïde. En revanche, la caspase-10, homologue de la caspase-8 [24, 25], pourrait jouer un rôle important étant donnée l’abondance de son ARNm dans la totalité des échantillons analysés.
Un point important de notre étude a été de comparer les données des biopuces concernant les populations cellulaires purifiées et celles d’échantillons tissulaires.
En effet, la fiabilité de l’analyse par biopuces sur tissu semble discutable, en raison de l’hétérogénéité intrinsèque du tissu tumoral, qui contient à la fois des cellules néoplasiques et des cellules réactionnelles du stroma. Notre étude montre que les concordances entre les profils d’expression des prélèvements testés et leurs caracté- ristiques histologiques ont été plus satisfaisantes pour les cellules purifiées que pour les tissus. Dans le premier groupe, les profils d’expression géniques permettaient de séparer nettement les lésions bénignes, les lymphomes folliculaires et les lymphomes diffus à grandes cellules, ce qui suggère une application possible des biopuces comme aide au diagnostic en hématopathologie. Ceci devra cependant être confirmé dans une étude portant sur une plus grande série.
Les progrès techniques constants concernant la miniaturisation des biopuces et les possibilités de la bio-informatique laissent espérer qu’une classification des LMNH à partir des données « moléculaires » pourrait, dans l’avenir, compléter les classifications actuelles en améliorant leur pouvoir prédictif de l’évolutivité de la maladie.
La mise en évidence d’une dérégulation de l’expression de certains gènes pourra aussi constituer une cible pour le développement de stratégies thérapeutiques nouvelles.
BIBLIOGRAPHIE [1] ZHAO N., HASHIDA H., TAKAHASHI N. et al . — High-density cDNA filter analysis : a novel approach for large-scale, quantitative analysis of gene expression.
Gene, 1995, 156, 207-213.
[2] LOCKART D.J., DONG H., BYRNE M.C. et al. — Expression monitoring by hybridization to high-density oligonucleotide arrays.
Nature Biotechnology , 1996, 14, 1675-1680.
[3] NGUYEN C., ROCHA D., GRANJEAUD S. et al . — Differential gene expression in the murine thymus assayed by quantitative hybridation of arrayed cDNA clones.
Genomics, 1995, 29, 207-216.
[4] TAKAHASHI N., HASHIDA H., ZHAO N. et al. — High-density cDNA filter analysis of the expression profiles of the genes preferentially expressed in human brain.
Gene, 1995, 164, 219-227.
[5] BERNARD K., AUPHAN N., GRANJEAUD S. et al. —Multiplex messenger assay : simultaneous, quantitative measurement of expression of many genes in the context of T cell activation.
Nucl.
Acid. Res., 1996, 24, 1435-1442.
[6] LOS M., WESSELBORG S., SCHULZE-OSHOFF K. — The role of caspases in development, immunity and apoptotic signal transduction : lessons from knock out mice. Immunity , 1999, 10 , 629-639.
[7] HARRIS N.L., JAFFE E.S., DIEBOLD J. et al. — World Health Organization classification of neoplasic diseases of the hematopoietic and lymphoid tissues : report of the Clinical Advisory Committee Meeting. Airlie House, Virginia, November 1997. J. Clin. Oncol., 1999, 17 , 3835- 3849.
[8] BAKHSHI A., JENSEN J.P., GOLDMAN P. et al . —Cloning the chromosomal break-point of t(14 ;
18) in human lymphomas : clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell, 1985, 41, 899-906.
[9] XERRI L., BOUABDALLAH R., DEVILARD E. et al. — Sensibility to Fas-mediated apoptosis is null or weak in B-cell non-Hodgkin’s lymphomas and is moderately increased by CD40 ligation.
Br.
J. Cancer , 1998, 78 , 225-232.
[10] PLUMAS J., JACOB M.C., CHAPEROT L., MOLENS J.P., SOTTO J.J., BENSA J.C. — Tumor B cells non-Hodgkin’s lymphoma are resistant to CD95 (FasApo-1)-mediated apoptosis. Blood, 1998, 91 , 2875-85.
[11] XERRI L., DEVILARD E., AYELLO C. et al. — Cysteine protease CPP32, but not Ich 1-L, is expressed in germinal center B cells and their neoplastic counterparts.
Human Pathol ., 1997, 28 , 912-21.
[12] KRAJEWSKA M., WANG H.G., KRAJEWSKRI S., ZAPATA J.M., SHABAIK A., GASCOYNE R., REED J.C. — Immunohistochemical analysis of in vivo pattern of expression of CPP32 (caspase-3), a cell death protease.
Cancer Res ., 1997, 57 , 1605-1613.
[13] XERRI L., DEVILARD E., BOUABDALLAH R., HASSOUN J., STOPPA A.M., BIRG F. — FADD expression and caspase activation in B-cell lymphomas resistant to Fas-mediated apoptosis. Br.
J. Haematol., 1999, 106 , 652-661.
[14] ALIZADEH A.A., EISEN M.B., DAVIS R.E. et al . — Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling.
Nature, 2000, 403 , 503-11.
[15] PENAULT-LLORCA F., BOUABDALLAH R., DEVILARD E. et al. — Analysis of bax expression in human tissues using the anti-bax 4F11 monoclonal antibody on paraffin sections . Pathol. Res.
Pract ., 1998, 194 , 457-64.
[16] WOLF B., SCHULER M., ECHEVERRI F., GREEN D. — Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/ inhibitor of caspaseactivated Dnase inactivation.
J. Biol. Chem ., 1999, 43 , 30651-30656.
[17] GERMAIN M., AFFAR B., D’AMOURS D., DIXIT V., SALVESEN G., POIRIER G. — Cleavage of automodified Poly(ADP-ribose)Polymerase during apoptosis . J. Biol. Chem ., 1999, 40 , 28379- 28384.
[18] DE CRAEN M.V., DECLERCQ W., DEN BRANDE I.V. — The proteolytic procaspase activation netwok : an in vitro analysis ? Cell Death Differ ., 1999, 6 , 1117-24.
[19] BERGERON L., PEREZ G.I., MACDONALD G. et al . — Defects in regulation of apoptosis in caspase-2-deficient mice.
Genes Dev ., 1998, 12 , 1304-14.
[20] WANG L., MIURA M., BERGERON L., ZHU H., YUAN J. — Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell , 1994, 78 , 739-50.
[21] FERNANDES-ALNEMRI T., ARMSTRONG R.C., KREBS J. et al . — In vitro activation of CPP32 and
Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natle Acad. Sci. USA , 1996, 93 , 7464-9.
[22] MUZIO M., CHINNAIYAN A.M., KISCHKEL F.C. et al . — FLICE, a novel FADD-homologous
ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell , 1996, 85 , 817-27.
[23] LI P., NIJHAWAN D., BUDIHARDJO I. et al . — Cytochrome c and dATP-dependent formation of
Apaf-1/caspase -9 complex initiates an apoptotic protease cascade.
Cell, 1997, 91, 479-89.
[24] NG P.W., PORTER A.G., JANICKE R.U. — Molecular cloning and characterization of two novel pro-apoptotic isoforms of caspase-10. J. Biol. Chem ., 1999, 274 , 10301-8.
[25] VINCENZ C., DIXIT V.M. — Fas-associated death domain protein interleukin-1 beta-converting enzyme 2 (FLICE2), an ICE/Ced-3 homologue, is proximally involved in CD95- and p55- mediated death signaling . J. Biol. Chem ., 1997, 272 , 6578-83.
DISCUSSION
M. Jacques-Louis BINET
Pourquoi avez-vous étudié si peu d’échantillons alors que votre Institut dispose d’un recrutement considérable ? Pourquoi aborder l’apoptose alors que celle-ci, dans les lymphomes, a une valeur pronostique très difficile à fixer (par exemple les Burkitt) ? Pourquoi, dans l’avenir, ne pas étudier par cette méthode, dans un même cadre histopathologique des membranes de haute-densité, les patients qui ont répondu ou non à un même traitement ?
Notre étude comporte relativement peu de patients car elle ne vise pas à rechercher d’éventuelles corrélations pronostiques qui auraient en effet exigé un effectif plus important. Le but de cette étude était avant tout de démontrer la faisabilité en routine d’une telle analyse, ce qui a nécessité une phase cruciale de mise au point des paramètres expérimentaux, des méthodes bio-informatiques de recueil des résultats et surtout de vérification de leur reproductibilité, une étape déterminante qui nécessite un processus complexe de normalisation à l’aide de témoins positifs. Une étude pronostique est actuellement en cours sur une plus grande série de patients.
M. Jean CIVATTE
Cette technique est-elle utile pour le diagnostic différentiel des lésions bénignes simulant tellement des lymphomes cutanés qu’on les appelle pseudo-lymphomes ?
Les « pseudo-lymphomes » cutanés représentent un exemple typique de lésion qui pourrait bénéficier de ce type d’analyse par biopuces ADN. En effet, ces lésions sont de pronostic incertain, avec une évolution possible vers un lymphome qui ne peut pas être prédite par l’analyse histologique conventionnelle. Il est donc nécessaire de rechercher de nouveaux marqueurs pronostiques, parmi lesquels les marqueurs moléculaires issus des profils transcriptionnels représentent une piste prometteuse pour l’avenir.
M. Maurice TUBIANA
L’étude du génome par ces techniques constitue certainement une des voies de recherche les plus intéressantes pour les lymphomes. Cependant, j’ai été frappé par la difficulté de ces recherches, sur le plan informatique et statistique. On se trouve en face d’une masse énorme de données avec un bruit de fond élevé et des variations relativement faibles autour de ce bruit. Ne pensez-vous pas qu’une étape préalable serait la mise au point de techniques d’analyse de ces données permettant d’identifier, au milieu de ces données, celles qui
pourraient être significatives ? Cette demande nécessite l’analyse d’un grand nombre de cas, donc des efforts conjoints de plusieurs équipes.
Les problèmes d’analyse bio-informatique sont, comme vous le soulignez, un écueil considérable en raison du flot de résultats générés par ces nouvelles techniques. C’est la raison pour laquelle une étape de mise au point de ces méthodes statistiques a été nécessaire, et ma réponse rejoint en celà celle que j’ai faite précédemment à M. le professeur Binet. Cependant, il faut bien réaliser que même avec des outils biostatistiques très perfectionnés, l’étude du transcriptome n’apportera pas toutes les réponses pour expliciter la cancérogenèse. En effet, l’analyse de l’ARNm ne permet pas de préjuger des phénomènes complexes d’interactions protéiques, qui jouent probablement aussi un rôle crucial dans la physiopathologie tumorale.
M. Raymond ARDAILLOU
Quel est l’intérêt d’un transcriptome total par rapport à la technique sélective que vous avez choisie ? L’utilisation du transcriptome total, sans définition préalable de gènes d’intérêt, permettrait de mettre en évidence des transcrits auxquels l’investigateur ne pense pas d’emblée.
Le terme transcriptome est en effet un abus de langage car aucune analyse par biopuces ADN ne peut étudier de façon exhaustive la totalité des gènes humains, ce qui serait d’ailleurs très délicat à gérer pour l’analyse des résultats. Une approche plus ciblée en sélectionnant d’emblée des gènes potentiellement impliqués dans les phénomènes étudiés est plus réaliste, notamment pour utiliser en pratique les données pour la prise en charge des patients.
* Département de pathologie ** INSERM U 119, Institut Paoli-Calmettes, IFR 57 et Université de la Méditerranée, Marseille. Tirés-à-part : Docteur Luc XERRI, Département de Pathologie, Institut Paoli-Calmettes, 232 Bld de Sainte Marguerite, BP 156-13273 Marseille cedex 9. Tél : +33 [0] 4 91223457. Fax : +33 [0] 4 91223573. Email : Article reçu le 20 juin 2000, accepté le 8 janvier 2001.
Bull. Acad. Natle Méd., 2001, 185, no 5, 963-975, séance du 29 mai 2001