
Résumé
Les pneumonies interstitielles fibrosantes regroupent différentes entités qui sont la pneumonie interstitielle commune, la pneumonie interstitielle non spécifique, la pneumonie interstitielle aiguë, la pneumonie interstitielle desquamative et la pneumonie interstitielle lymphocytaire. Du fait d’un pronostic plus péjoratif et d’une prise en charge différente, la pneumonie interstitielle commune surtout quand elle est idiopathique (fibrose pulmonaire idiopathique) doit être différenciée des autres pneumonies interstitielles. Le diagnostic de fibrose pulmonaire idiopathique est basé sur des critères scanographiques ou histologiques de pneumonie interstitielle commune, en l’absence d’asbestose, de pneumonie d’hypersensibilité chronique et de collagénoses. Dans plus de 50 % des cas, un diagnostic de fibrose pulmonaire idiopathique peut être fait sur la base des signes scanographiques (opacités réticulées, rayon de miel de distribution prédominante basale et sous-pleurale), en l’absence de signe atypique (nodules, condensations parenchymateuses ou verre dépoli étendue). En l’absence de ces critères et en raison d’un recouvrement important des signes radiologiques des différentes pneumonies interstitielles fibrosantes, la biopsie pulmonaire peut être requise. Le diagnostic repose alors sur la confrontation entre les données cliniques, radiologiques et histopathologiques.
Summary
The term fibrosing interstitial pneumonia covers several distinct entities, including usual interstitial pneumonia, non specific interstitial pneumonia acute interstitial pneumonia, desquamative interstitial pneumonia, and lymphoid interstitial pneumonia. Because of its very poor prognosis and different management, usual interstitial pneumonia, and particularly idiopathic forms (idiopathic pulmonary fibrosis), must be distinguished from other forms of interstitial pneumonia. The diagnosis of idiopathic pulmonary fibrosis is based on the CT or pathologic criteria of usual interstitial pneumonia, in the absence of asbestosis, chronic hypersensitive pneumonitis, and collagen vascular disease. In more than 50 % of cases, idiopathic pulmonary fibrosis may be confidently diagnosed on the basis of CT findings, showing reticular opacities and honeycombing with a predominantly basal and subpleural distribution, without nodules, extensive consolidation or ground-glass opacities. Surgical biopsy may be necessary when these features are absent, given the overlap of CT findings between the different forms of interstitial pneumonia. In such cases, specific diagnosis of interstitial lung disease is based on a combination of clinical, radiological and histopathological findings.
Les pneumonies interstitielles fibrosantes regroupent des entités différentes qui n’ont pas le même pronostic. Le diagnostic de ces différentes entités est basé sur la confrontation des signes cliniques, radiologiques et anatomopathologiques [1, 2]. Le rôle du clinicien est de déterminer si l’atteinte pulmonaire est idiopathique ou liée à une exposition à des particules inhalées fibrogènes ou des antigènes allergisants, ou associée à une collagénose, ou d’origine médicamenteuse. Le rôle du radiologiste est de déterminer si les manifestations radiologiques de l’atteinte pulmonaire sont caractéristiques d’une pneumonie interstitielle commune, qu’elle soit liée à une cause identifiée ou idiopathique (fibrose pulmonaire idiopathique) ou si les signes sont non spécifiques. La décision de réaliser une biopsie pulmonaire est alors basée sur l’analyse conjointe du clinicien et du radiologiste. L’objectif de cet article est de revoir les signes radiologiques des différentes pneumonies interstitielles fibrosantes et de préciser le rôle du radiologiste dans la stratégie diagnostique de ces maladies.
Fibrose pulmonaire idiopathique (FIP)
La FIP est définie comme une forme de pneumonie interstitielle fibrosante chronique strictement limitée aux poumons et associée à un aspect anatomopathologique de pneumonie interstitielle commune (PIC). La FIP est la plus fréquente des pneumonies interstitielles idiopathiques. Elle est même plus fréquente chez les fumeurs que chez les non-fumeurs. Des formes familiales peuvent être occasionnellement rencontrées. Bien que normale dans environ 10 % des cas, la radiographie du thorax montre le plus souvent des opacités pulmonaires de type interstitiel au niveau des bases : petites opacités linéaires, irrégulières, responsables d’un aspect réticulé bilatéral souvent symétrique. Les lésions peuvent être diffuses et prédominer aux bases ou siéger exclusivement dans les bases pulmonaires. Lorsque la maladie s’aggrave, les anomalies radiographiques sont de plus en plus étendues donnant un aspect de réticulations plus grossières, voire un aspect réticulonodulaire avec une perte progressive du volume pulmonaire. Dans les cas les plus avancés, l’aspect devient caractéristique, fait d’images en rayon de miel dans lesquelles les kystes mesurent entre 0,5 et 10 mm.
L’aspect scanographique est relativement caractéristique de la maladie, fait de plages multiples de réticulations intralobulaires siégeant dans les territoires sous-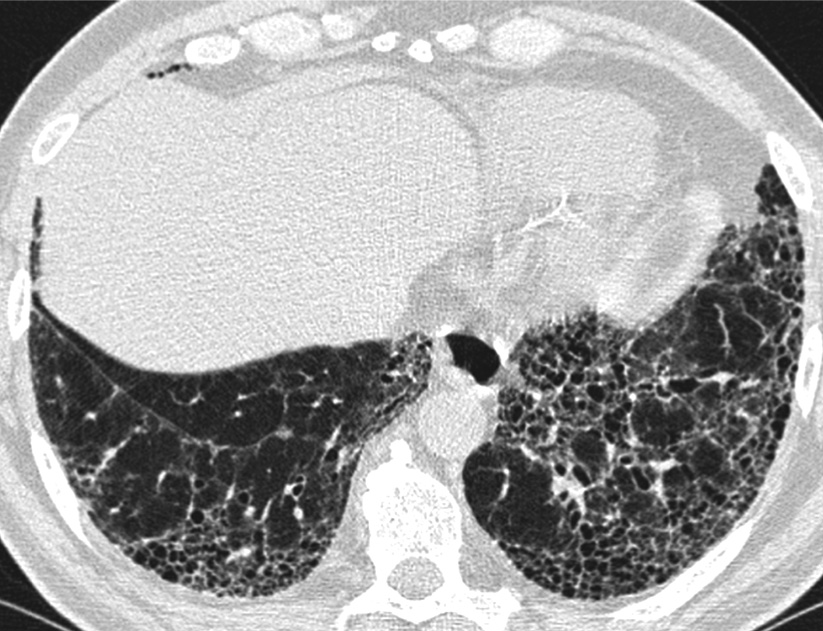 pleuraux, en particulier dans les zones inférieures des poumons. Ces réticulations sont associées à une distorsion de l’architecture pulmonaire avec dilatations variqueuses des bronches et des bronchioles (bronchectasies et bronchiolectasies par traction) et irrégularités des interfaces entre le poumon et la plèvre, les vaisseaux et les bronches. Des images kystiques en couches jointives, siégeant dans les territoires périphériques sous-pleuraux des poumons, sont présentes dans 90 % des cas (rayon de miel) [3]. Dans la très grande majorité des cas de FPI, la distribution des réticulations intralobulaires et du rayon de miel est de type sous-pleural et la fibrose semble plus sévère dans les zones inférieures des poumons (Fig. 1). Malgré cette prédominance de la sévérité dans les territoires inférieurs, les réticulations et le rayon de miel peuvent toucher toutes les zones pulmonaires, avec toutefois une hétérogénéité de distribution, les zones de fibrose alternant avec des zones de parenchyme apparaissant normales. Des opacités en verre dépoli sont fréquemment présentes, mais toujours discrètes et associées à des signes évidents de fibrose pulmonaire. Des zones de verre dépoli visibles en dehors des zones de fibrose sont aussi présentes dans environ un quart des cas mais leur étendue est inférieure à celle des réticulations. Ce verre dépoli peut refléter soit la présence d’une inflammation active, soit la présence d’une fibrose microscopique [4] . Le verre dépoli dû à une fibrose microscopique est souvent associé à des réticulations, des bronchectasies et des bronchiolectasies par traction. De façon tout à fait occasionnelle, des petites calcifications de forme linéaire ou nodulaire sont vues dans les zones de fibrose. Elles reflètent un phénomène d’ossification. Une hypertrophie modérée des ganglions lymphatiques médiastinaux est notée dans environ 70 % des cas [5]. Chez les patients fumeurs, des signes scanographiques de BPCO peuvent être associés aux signes de la fibrose, l’emphysème prédominant dans les lobes supérieurs plus ou moins associé à des zones d’hypodensité avec perfusion en mosaïque et quelques petits nodules centrolobulaires.
pleuraux, en particulier dans les zones inférieures des poumons. Ces réticulations sont associées à une distorsion de l’architecture pulmonaire avec dilatations variqueuses des bronches et des bronchioles (bronchectasies et bronchiolectasies par traction) et irrégularités des interfaces entre le poumon et la plèvre, les vaisseaux et les bronches. Des images kystiques en couches jointives, siégeant dans les territoires périphériques sous-pleuraux des poumons, sont présentes dans 90 % des cas (rayon de miel) [3]. Dans la très grande majorité des cas de FPI, la distribution des réticulations intralobulaires et du rayon de miel est de type sous-pleural et la fibrose semble plus sévère dans les zones inférieures des poumons (Fig. 1). Malgré cette prédominance de la sévérité dans les territoires inférieurs, les réticulations et le rayon de miel peuvent toucher toutes les zones pulmonaires, avec toutefois une hétérogénéité de distribution, les zones de fibrose alternant avec des zones de parenchyme apparaissant normales. Des opacités en verre dépoli sont fréquemment présentes, mais toujours discrètes et associées à des signes évidents de fibrose pulmonaire. Des zones de verre dépoli visibles en dehors des zones de fibrose sont aussi présentes dans environ un quart des cas mais leur étendue est inférieure à celle des réticulations. Ce verre dépoli peut refléter soit la présence d’une inflammation active, soit la présence d’une fibrose microscopique [4] . Le verre dépoli dû à une fibrose microscopique est souvent associé à des réticulations, des bronchectasies et des bronchiolectasies par traction. De façon tout à fait occasionnelle, des petites calcifications de forme linéaire ou nodulaire sont vues dans les zones de fibrose. Elles reflètent un phénomène d’ossification. Une hypertrophie modérée des ganglions lymphatiques médiastinaux est notée dans environ 70 % des cas [5]. Chez les patients fumeurs, des signes scanographiques de BPCO peuvent être associés aux signes de la fibrose, l’emphysème prédominant dans les lobes supérieurs plus ou moins associé à des zones d’hypodensité avec perfusion en mosaïque et quelques petits nodules centrolobulaires.
Fig. 1. — Fibrose pulmonaire idiopathique. Coupe scanographique de 1 mm passant par les bases pulmonaires. Réticulations intralobulaires et rayon de miel de topographie prédominante souspleurale. Bronchectasies et bronchiolectasies par traction.
Au cours du suivi évolutif de la maladie, les zones de verre dépoli peuvent soit régresser, soit plus fréquemment se transformer en réticulations ou rayon de miel.
L’évolution se caractérise par une aggravation inexorable et progressive aussi bien en étendue qu’en sévérité de la fibrose. Quelques zones de verre dépoli peuvent régresser pendant que d’autres sont remplacées par des réticulations et du rayon de miel. Les réticulations se transforment souvent en rayon de miel et les kystes de rayon de miel augmentent progressivement en taille. Lors des poussées aiguës de la maladie, les examens scanographiques peuvent montrer des zones de verre dépoli ou de condensations parenchymateuses superposées aux zones de réticulations et de rayons de miel. Ces condensations parenchymateuses peuvent être diffuses, multifocales ou périphériques. Elles doivent faire suspecter soit une détérioration clinique aiguë de la maladie, soit une infection opportuniste, en particulier une pneumonie à pneumocystis jiroveci [4].
Pneumonie interstitielle non spécifique (PINS)
Il s’agit d’une pneumonie interstitielle chronique caractérisée par un épaississement homogène des parois alvéolaires par une réaction inflammatoire ou de la fibrose, ou souvent l’association des deux. La PINS est la plus fréquente des pneumonies interstitielles chroniques après la fibrose pulmonaire idiopathique. La PINS peut être idiopathique mais le plus souvent se voit comme la manifestation d’une collagénose, d’une pneumonie d’hypersensibilité, d’une pneumonie d’origine médicamenteuse ou comme une maladie interstitielle chronique compliquant un dommage alvéolaire diffus. Le pronostic de la PINS est influencé par la composante histologique prédominante. Les patients avec une composante prédominante ou exclusive inflammatoire (PINS cellulaire) ont un excellent pronostic, tandis que la survie moyenne des patients avec une PINS fibrosante varie entre 6 et 14 ans. Le pronostic de la PINS est significativement meilleur que celui de la fibrose pulmonaire idiopathique.
Les manifestations radiographiques les plus fréquentes de la PINS sont des zones d’augmentation de la densité pulmonaire (verre dépoli) touchant principalement les zones moyennes et inférieures des deux poumons, distribuées de façon bilatérale en plages plus ou moins confluentes. Les autres signes sont des réticulations ou une association de réticulations de verre dépoli et de condensations parenchymateuses.
La radiographie pulmonaire est normale chez 15 % des patients atteints de PINS.
Sur les examens scanographiques, l’infiltration interstitielle pulmonaire est toujours visible. L’aspect le plus fréquemment rencontré se caractérise par des opacités en verre dépoli bilatérales et symétriques [1-3, 6]. Chez la plupart des patients, il existe aussi de fines réticulations intralobulaires et des bronchectasies par traction superposées aux zones de verre dépoli (Fig. 2). Le rayon de miel n’est rencontré que dans 10 à 30 % des cas, et quand il est présent reste limité à moins de 10 % du territoire pulmonaire. Quelques plages de condensations parenchymateuses peuvent être vues. Elles reflètent les zones de pneumonie organisée focales, essentiellement chez les patients atteints de collagénose.
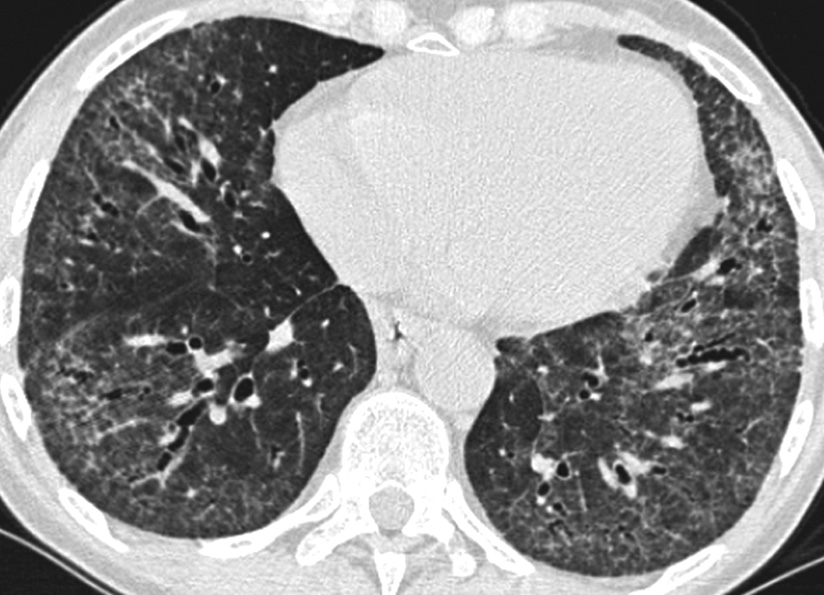
Fig. 2. — Pneumonie interstitielle non spécifique. Plages bilatérales et symétriques de verre dépoli contenant de fines réticulations intralobulaires et des bronchectasies par traction.
Les anomalies scanographiques au cours de la PINS sont très souvent diffuses, mais prédominent dans plus de la moitié des cas dans les territoires inférieurs des poumons et principalement dans les zones périphériques. Bien que la distribution soit fréquemment périphérique, il a été démontré que chez environ 50 % des patients avec PINS fibrosante, cette fibrose respecte le parenchyme pulmonaire immédiatement adjacent à la plèvre sur au moins deux niveaux au sein des lobes inférieurs [7].
Ce respect relatif des zones sous-pleurales peut être utilisé de façon positive pour différencier les PINS avec fibrose de la pneumonie interstitielle commune. Les patients atteints de PINS qui présentent des opacités en verre dépoli sans réticulations et sans modification des voies aériennes ont typiquement une PINS cellulaire.
Les patients avec verre dépoli associé à des réticulations et des bronchectasies par traction peuvent avoir soit une PINS cellulaire soit une PINS fibrosante [6, 8, 9].
Une hypertrophie des ganglions médiastinaux est scanographiquement évidente chez 80 % des patients atteints de PINS [5]. Cette hypertrophie est habituellement modérée (les ganglions mesurent entre 10 et 15 mm dans leur petit axe).
Le suivi évolutif des patients atteints de PINS a montré que ceux avec verre dépoli prédominant sur le scanner initial avaient tendance à s’améliorer sous traitement avec un meilleur pronostic à long terme que les patients avec signes scanographiques de fibrose prédominants. L’aspect scanographique initial des poumons des patients atteints de PINS est significativement associé aux modifications ultérieures sur les examens de surveillance. Ainsi dans une étude, tous les patients présentant des lésions inflammatoires prédominantes sur l’examen scanographique initial montraient après un an d’évolution des signes d’amélioration tandis que ceux ayant une atteinte initiale de fibrose prédominante s’amélioraient, se détérioraient ou restaient stables [10]. Inversement dans cette même étude, il n’y avait pas d’association significative entre les signes histologiques et la prédiction d’une amélioration sur les examens ultérieurs.
Comme chez les patients atteints de FPI, ceux atteints de PINS peuvent présenter une détérioration aiguë avec une aggravation brutale des symptômes liée à une infection, une embolie pulmonaire ou une insuffisance cardiaque [4, 11]. Quand aucune cause de cette aggravation aiguë n’est identifiée, on parle de phase d’accélé- ration de la maladie. Une telle accélération survient dans environ 3 à 4 % des patients [12]. L’atteinte histologique consiste en une pneumonie organisée ou plus fréquemment un dommage alvéolaire diffus et les signes scanographiques consistent en des plages extensives de verre dépoli et de condensations parenchymateuses superposées aux réticulations [13].
Pneumonie interstitielle aiguë (PIA)
La PIA est une maladie aiguë sévère de cause inconnue qui survient chez un sujet préalablement sain et qui produit des anomalies histologiques de dommage alvéolaire diffus. Les manifestations radiologiques sont identiques à celles vues au cours du syndrome de détresse respiratoire aigu (SDRA) [1]. La différence n’est qu’aucune étiologie dans ce cas n’est retrouvée. La PIA est en fait un SDRA idiopathique. Les manifestations radiographiques sont similaires à celles d’un SDRA et consistent en des zones de condensations parenchymateuses bilatérales avec bronchogramme aérique. Les condensations sont initialement en plages dispersées, mais celles-ci deviennent rapidement confluentes et diffuses bien qu’elles aient une prédominance dans les parties supérieures ou inférieures du poumon. Le volume pulmonaire est habituellement diminué [14].
En scanographie, les manifestations les plus précoces de la PIA sont des plages de verre dépoli bilatérales ou d’un verre dépoli diffus [3]. La plupart des patients ont un discret épaississement régulier des septa interlobulaires et des lignes intralobulaires superposées au verre dépoli responsable d’un aspect dit en « crazy paving ». Le respect de plusieurs lobules pulmonaires isolés au sein du verre dépoli donne à l’ensemble un aspect typique en carte de géographie [3 , 15]. Les condensations parenchymateuses sont présentes chez la plupart des patients. Elles sont volontiers en plages, dispersées ou confluentes, et ont tendance à se localiser dans les territoires déclives. Quand la maladie s’aggrave, les plages en verre dépoli deviennent diffuses, les zones de condensation deviennent plus étendues et une distorsion architecturale et des bronchectasies par traction deviennent apparentes [3, 15]. Le rayon de miel est présent dans 10 à 26 % des cas. Les autres signes associés sont des petits épanchements pleuraux vus dans 30 % des cas et une hypertrophie modérée des ganglions médiastinaux dans 5 à 10 % des cas [14, 16]. Des corrélations anatomoradiologiques ont montré que les zones de verre dépoli et les condensations parenchymateuses sans bronchectasies par traction reflétaient les phases exsudative et proliférative de la PIA tandis que les bronchectasies par traction n’étaient vues que dans les phases prolifératives et fibreuses de la maladie. Les survivants peuvent présenter des réticulations périphériques modérées, des bronchectasies et des bronchiolectasies par traction et du rayon de miel.
La pneumonie d’hypersensibilité (PHS)
La PHS ou alvéolite allergique extrinsèque est une pneumonie interstitielle diffuse caractérisée par une réaction inflammatoire immuno-induite due à une inhalation d’antigènes qui affecte certains patients. La liste des antigènes inhalés potentiellement responsables est vaste. Une réaction du même type peut aussi survenir avec certains médicaments.
Quel que soit l’antigène responsable, les manifestations radiographiques sont les mêmes [17]. A la phase aiguë, l’aspect est celui d’opacités alvéolaires dont la distribution est similaire à celle de l’œdème pulmonaire aigu. Des micronodules à contours flous peuvent aussi être détectés. A la phase subaiguë, l’aspect est fait de plages mal limitées de verre dépoli et d’opacités nodulaires diffuses à contours mal définis. A la phase chronique, il s’agit d’opacités linéaires irrégulières et d’images en rayon de miel avec perte de volume pulmonaire. La fibrose est diffuse ou prédomine dans les parties supérieure, moyenne ou inférieure. Des adénopathies hilaires et médiastinales sont souvent détectées.
En scanographie les phases aiguë et subaiguë sont caractérisées par des plages de condensations parenchymateuses et de verre dépoli associées à des micronodules centrolobulaires à contours flous [18]. D’autres signes font partie du tableau, en particulier des zones lobulaires hypodenses et hypovascularisées avec piégeage expiratoire exprimant de façon indirecte l’obstruction bronchiolaire par la bronchiolite cellulaire ou la bronchiolite oblitérative, mais aussi des nodules irréguliers mesurant plus de 10 mm représentant des zones focales de pneumonie organisée et des kystes pulmonaires à parois fines [19]. Au stade chronique de la maladie, l’expression scanographique est faite de signes de fibrose associant réticulations, distorsion architecturale, bronchectasies et bronchiolectasies par traction, et rayon de miel mimant ainsi l’aspect d’une FPI [18, 20]. Le rayon de miel est toujours minime et a rarement une distribution basale. La distribution de la fibrose est extrêmement variable soit moyenne respectant les apex et les bases, soit prédominante aux lobes supérieurs ou aux lobes inférieurs, soit diffuse. Dans la plupart des cas, les signes de pneumonie subaiguë sont aussi présents, superposés aux signes de fibrose (Fig. 3). La plupart des cas présentent des plages de verre dépoli. Des micronodules centrolobulaires sont vus dans 50 % des cas permettant de suggérer le diagnostic de PHS. Des zones lobulaires de piégeage expiratoire sont rapportées dans 80 % des cas et les kystes dans 40 % [20]. Des adénopathies médiastinales sont fréquentes.
Pneumonie interstitielle desquamative (PID)
La PID est une entité rare qui est fortement liée au tabagisme. La lésion histopathologique de la PID est un remplissage alvéolaire diffus par des macrophages plus ou moins associés à un épaississement minime des septa alvéolaires par de cellules alvéolaires et de la fibrose. Bien que la PID soit souvent classée comme une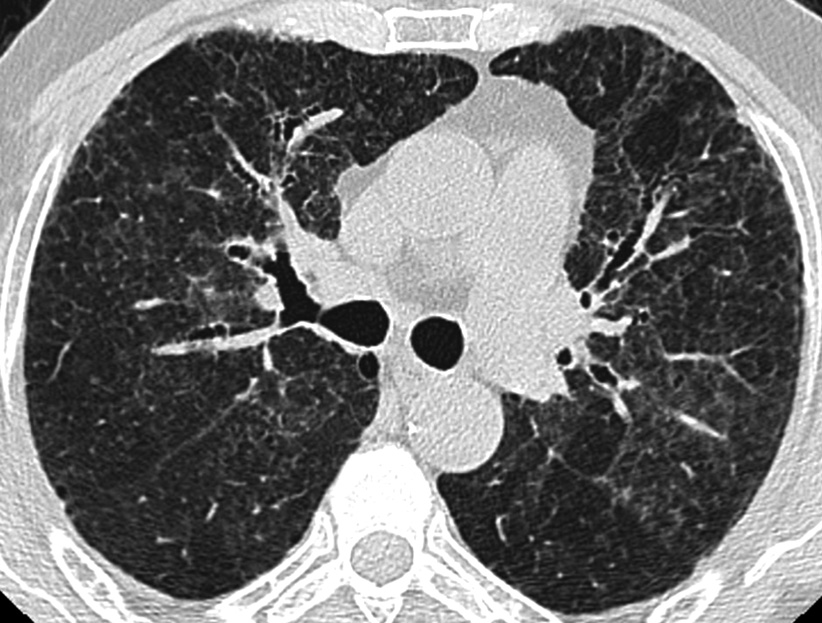
Fig. 3. — Pneumonie d’hypersensibilité chronique. Plages bilatérales de verre dépoli contenant des micronodules centrolobulaires et de fines réticulations ; bronchectasies par traction sans rayon de miel ; zones lobulaires apparaissant hypodenses et hypovascularisées dans les territoires antérieurs.
pneumonie interstitielle idiopathique, plus de 90 % des patients atteints de PID sont des fumeurs et cette maladie est sensée représenter une partie du spectre histopathologique des maladies interstitielles pulmonaires liées à la fumée de cigarette incluant l’histiocytose à cellules de Langerhans et la bronchiolite respiratoire associée à une maladie interstitielle pulmonaire [21]. De plus, un aspect histopathologique tout à fait voisin peut être associé à certaines pneumonies par inhalation de poussières, certaines réactions médicamenteuses et des collagénoses.
Les manifestations radiographiques de la PID consistent en un verre dépoli bilatéral et une diminution du volume des deux poumons. Les autres signes rapportés sont non spécifiques, opacités réticulées ou réticulonodulaires avec une distribution bibasale. Les radiographies du thorax peuvent quelquefois apparaître normales [1].
Sur les examens scanographiques, le signe majeur est le verre dépoli qui est diffus avec une distribution prédominante sous-pleurale dans les zones inférieures du poumon [3]. Une discrète réticulation intralobulaire est présente dans environ 50 % des cas et quelques petites zones de rayon de miel sont visibles dans un petit pourcentage de cas. La distorsion architecturale des poumons et les bronchectasies par traction sont des signes très inhabituels. Dans de nombreux cas, de petits espaces kystiques dans les zones de verre dépoli sont visibles. Ils correspondent vraisemblablement à des petites bronchiolectasies et des dilatations des canaux alvéolaires. Ces kystes peuvent disparaître spontanément sans fibrose en rayon de miel. A noter la présence fréquente d’emphysème centrolobulaire et paraseptal au sein des zones de verre dépoli qu’il peut être difficile de différencier des kystes pulmonaires.
Les examens scanographiques de surveillance de la maladie ont montré que les zones de verre dépoli tendaient à rester stables au cours du temps ou s’amélioraient sous corticoïdes. Quelques patients peuvent présenter une aggravation de la maladie avec augmentation de l’étendue du verre dépoli en dépit des corticoïdes [22].
Pneumonie interstitielle lymphocytaire (PIL)
La PIL est une entité rare caractérisée en anatomopathologie par une infiltration diffuse des septa alvéolaires par des lymphocytes polyclonaux. La plupart des cas de PIL se voient chez des patients ayant une maladie autoimmune ou un état d’immunodéficience. La PIL survient le plus souvent au cours des syndromes de Sjögren, d’une maladie autoimmune, au cours de certaines dysprotéinémies, ou au cours du SIDA particulièrement chez l’enfant. Les PIL dites idiopathiques sont extrêmement rares.
Sur le plan radiographique, l’aspect est celui d’opacités réticulaires ou réticulonodulaires infiltrant principalement les parties inférieures des poumons. De façon moins fréquente, un syndrome nodulaire, des opacités floues en verre dépoli ou des condensations parenchymateuses sont présentes. Des lésions kystiques peuvent aussi se voir.
En scanographie, l’atteinte consiste en un verre dépoli bilatéral, extensif et des nodules centrolobulaires à contours mal définis [3]. Des kystes pulmonaires peuvent être présents. Ces kystes sont la conséquence d’une obstruction luminale partielle des bronchioles par l’infiltration cellulaire péribronchiolaire. Ils mesurent 1 à 30 mm de diamètre et ont des parois fines et une distribution prédominante en périvasculaire ou sous-pleural. Ils n’occupent généralement pas plus de 10 % du parenchyme pulmonaire [23, 24]. D’autres manifestations moins fréquentes peuvent être rencontrées : des nodules mesurant de 1 à 2 cm de diamètre, des bronchectasies par traction et quelques plages en rayon de miel. Une discrète hypertrophie des ganglions médiastinaux est fréquemment présente. Occasionnellement, des nodules sont pré- sents au contact ou au sein des kystes pulmonaire reflétant la présence de dépôts amyloïdes qui peuvent se calcifier.
Sur les examens de surveillance, le verre dépoli et les petites opacités nodulaires centrolobulaires régressent progressivement alors que les kystes habituellement persistent et peuvent augmenter en taille. Des images de rayon de miel et de fibrose pulmonaire peuvent se développer chez quelques patients là où préalablement se trouvaient des plages de condensations parenchymateuses ou de verre dépoli.
Diagnostic différentiel
Le diagnostic des FPI peut être fait en l’absence de confirmation histologique chez de nombreux patients. Un diagnostic de FPI peut être établi sur des critères cliniques, fonctionnels et scanographiques chez 50 à 70 % des patients avec une spécificité supérieure à 90 % [25, 26]. La biopsie pulmonaire chirurgicale devrait être réservée aux patients chez lesquels les signes cliniques et/ou radiologiques de la maladie sont atypiques. L’asbestose peut être facilement différenciée de la FPI par la présence de plaques pleurales ou d’épaississements pleuraux diffus [27].
Les entités qui ont fréquemment une apparence similaire à la FPI en scanographie haute résolution sont la PINS et la PHS chronique. La différence entre ces trois entités est importante car leur prise en charge est totalement différente. La pneumonie interstitielle qui mime le plus fréquemment la FPI est la PINS qu’elle soit idiopathique ou associée aux connectivites [28, 29]. Un diagnostic scanographique confiant de FPI nécessite l’exclusion clinique des causes connues de pneumonies interstitielles communes et la présence des trois critères scanographiques : — présence de réticulations intralobulaires avec une distribution prédominante périphérique et basale, — présence de rayon de miel avec une distribution prédominante périphérique et basale, — absence de signes atypiques tels que nodules centrolobulaires, nodules péribronchovasculaires et condensations parenchymateuses étendues ou verre dépoli étendu. Si seulement les premier et troisième critères sont présents (rayon miel absent), les signes peuvent être interprétés comme une FPI probable. Les signes les plus prédictifs de la FPI sur les examens scanographiques haute résolution sont le rayon de miel dans les zones inférieures des poumons et les réticulations intralobulaires dans les parties supérieures [30]. Bien que le rayon de miel périphé- rique et basal soit un fort élément prédictif de FPI, il faut savoir le reconnaître de façon fiable. La reconnaissance du signe en scanographie nécessite la présence d’amas de petits éléments aériques et kystiques mesurant de 2 mm à 1 cm de diamètre, ayant des parois bien définies et épaisses et localisés au contact de la plèvre. Ils doivent être différenciés des images de bronchiolectasies par traction.
Celles-ci peuvent avoir une apparence similaire sur une coupe mais sont typiquement localisées à quelques millimètres ou plus de la plèvre.
Les réticulations intralobulaires et le rayon de miel sont aussi des signes fréquemment vus au cours de la PHS chronique [18]. La différence entre la FPI et la PHS chronique peut être faite sur la présence de nodules centrolobulaires, de zones lobulaires d’hypoatténuation et d’hypoperfusion et le respect relatif des bases pulmonaires chez les patients atteints de PHS. Silva et coll . ont vu les signes scanographiques d’une série de patients ayant une FPI, une PINS ou une PHS chronique [20]. Un diagnostic spécifique d’une de ces entités a pu être fait avec un haut niveau de confiance chez 53 % des cas, et le diagnostic proposé était correct dans 94 % des cas. La distinction entre ces trois maladies peut être faite sur la base des signes scanographiques dans approximativement 50 % des cas. Si l’on joint l’histoire clinique, les signes scanographiques permettent d’éviter la biopsie pulmonaire dans un très grand nombre de cas.
BIBLIOGRAPHIE [1] American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. , 2000, 161 , 646-664.
[2] Cottin V., Capron F., Grenier P., Cordier J.F. — [Diffuse idiopathic interstitial pneumonias.
International multidisciplinary consensus classification by the American Thoracic Society and the European Respiratory Society, principal clinico-pathological entities, and diagnosis].
Rev.
Mal Respir. , 2004, 21 , 299-318.
[3] Lynch D.A., Travis W.D., Muller N.L. et al. — Idiopathic interstitial pneumonias: CT features.
Radiology , 2005, 236 , 10-21.
[4] Silva C.I., Muller N.L., Fujimoto K. et al. — Acute exacerbation of chronic interstitial pneumonia: high-resolution computed tomography and pathologic findings.
J. Thorac. Imaging , 2007, 22 , 221-229.
[5] Souza C.A., Muller N.L., Lee K.S. et al. — Idiopathic interstitial pneumonias: prevalence of mediastinal lymph node enlargement in 206 patients.
AJR Am. J. Roentgenol. , 2006, 186 , 995-999.
[6] Johkoh T., Muller N.L., Colby T.V. et al. — Nonspecific interstitial pneumonia: correlation between thin-section CT findings and pathologic subgroups in 55 patients.
Radiology , 2002, 225 , 199-204.
[7] Silva C.I., Muller N.L., Hansell D.M. et al . — Nonspecific interstitial pneumonia and idiopathic pulmonary fibrosis: changes in pattern and distribution of disease over time.
Radiology , 2008, 247 , 251-259.
[8] Sumikawa H., Johkoh T., Ichikado K. et al . — Usual interstitial pneumonia and chronic idiopathic interstitial pneumonia: analysis of CT appearance in 92 patients.
Radiology , 2006, 241 , 258-266.
[9] Tsubamoto M., Muller N.L., Johkoh T. et al . — Pathologic subgroups of nonspecific interstitial pneumonia: differential diagnosis from other idiopathic interstitial pneumonias on high-resolution computed tomography. J. Comput. Assist. Tomogr. , 2005, 29 , 793-800.
[10] Screaton N.J., Hiorns M.P., Lee K.S. et al. — Serial high resolution CT in non-specific interstitial pneumonia: prognostic value of the initial pattern.
Clin. Radiol. , 2005, 60 , 96-104.
[11] Churg A., Muller N.L., Silva C.I., Wright J.L. — Acute exacerbation (acute lung injury of unknown cause) in UIP and other forms of fibrotic interstitial pneumonias. Am. J. Surg. Pathol. , 2007, 31 , 277-284.
[12] Park I.N., Kim D.S., Shim T.S. et al . — Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis.
Chest , 2007, 132 , 214-220.
[13] Kim D.S., Park J.H., Park B.K. et al . — Acute exacerbation of idiopathic pulmonary fibrosis:
frequency and clinical features.
Eur. Respir. J. , 2006, 27 , 143-150.
[14] Primack S.L., Hartman T.E., Ikezoe J. et al. — Acute interstitial pneumonia: radiographic and CT findings in nine patients.
Radiology , 1993, 188 , 817-820.
[15] Ichikado K., Suga M., Muller N.L. et al . — Acute interstitial pneumonia: comparison of high-resolution computed tomography findings between survivors and nonsurvivors.
Am J Respir Crit Care Med , 2002, 165 , 1551-1556.
[16] Johkoh T., Muller N.L., Taniguchi H. et al . — Acute interstitial pneumonia: thin-section CT findings in 36 patients.
Radiology , 1999, 211 , 859-863.
[17] Adler B.D., Padley S.P., Muller N.L., Rémy-Jardin M., Rémy J. — Chronic hypersensitivity pneumonitis: high-resolution CT and radiographic features in 16 patients. Radiology , 1992, 185 , 91-95.
[18] Silva C.I., Churg A., Muller N.L. — Hypersensitivity pneumonitis: spectrum of highresolution CT and pathologic findings. AJR Am. J. Roentgenol. , 2007, 188 , 334-344.
[19] Franquet T., Hansell D.M., Senbanjo T., Remy-Jardin M., Muller N.L. — Lung cysts in subacute hypersensitivity pneumonitis. J. Comput. Assist. Tomogr. , 2003, 27 , 475-478.
[20] Silva C.I., Muller N.L., Lynch D.A. et al. — Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology , 2008, 246 , 288-297.
[21] Ryu J.H., Colby T.V., Hartman T.E., Vassalo R. — Smoking-related interstitial lung diseases: a concise review. Eur. Respir. J. , 2001, 17 , 122-132.
[22] Akira M., Yamamoto S., Hara H., Sakatani M., Ueda E. — Serial computed tomographic evaluation in desquamative interstitial pneumonia. Thorax , 1997, 52 , 333-337.
[23] Johkoh T., Ichikado K., Akira M. et al. — Lymphocytic interstitial pneumonia: follow-up CT findings in 14 patients.
J. Thorac. Imaging , 2000, 15 , 162-167.
[24] Silva C.I., Flint J.D., Levy R.D., Miller N.L. — Diffuse lung cysts in lymphoid interstitial pneumonia: high-resolution CT and pathologic findings. J. Thorac. Imaging , 2006, 21 , 241-244.
[25] Hunninghake G.W., Zimmerman M.B., Schwartz D.A. et al . — Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis.
Am. J. Respir. Crit. Care Med. , 2001, 164 , 193-196.
[26] Raghu G., Mageto Y.N., Lockhart D. et al . — The accuracy of the clinical diagnosis of new-onset idiopathic pulmonary fibrosis and other interstitial lung disease: A prospective study.
Chest , 1999, 116 , 1168-1174.
[27] Copley S.J., Wells A.U., Sivakumaran P. et al. — Asbestosis and idiopathic pulmonary fibrosis: comparison of thin-section CT features.
Radiology , 2003, 229 , 731-736.
[28] Elliot T.L., Lynch D.A., Newell J.D. JR. et al. — High-resolution computed tomography features of nonspecific interstitial pneumonia and usual interstitial pneumonia.
J Comput Assist Tomogr , 2005, 29 , 339-345.
[29] MacDonald S.L., Rubens M.B., Hansell D.M. et al. — Nonspecific interstitial pneumonia and usual interstitial pneumonia: comparative appearances at and diagnostic accuracy of thin-section CT. Radiology , 2001, 221 , 600-605.
[30] Hunninghake G.W., Lynch D.A., Galvin J.R. et al. — Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia.
Chest , 2003, 124 , 1215-1223.
DISCUSSION
M. Emmanuel-Alain CABANIS
Pour l’inflammation, sa néoangiogenèse s’accompagne d’une modification locale, analysée par le scanner à rayons × avant et après injection iodée. Pour quelle raison n’avez vous pas évoqué la densitométrie spontanée, sans injection de contraste ? Votre expression « verre dépoli » évoque la séméiologie radiologique d’autrefois. Or, par définition, la densitométrie exprime une densité locale, bien sûr variable avec la technique (épaisseur des coupes, rythme cardiaque, vs balayage spirale…). A quelles valeurs de densité, en pourcentage relatif seulement, bien sûr, correspond ce « verre dépoli » de la FPI ? En matière pronostique et surveillance thérapeutique, même brève et sans acutisation clinique, quelle est la part et l’indication du scan Rx et à quel rythme ?
En routine clinique nous ne mesurons pas la densité pulmonaire, nous nous contentons de l’évaluer subjectivement. Les expressions poumon de densité normale, hyperdensité en verre dépoli et condensation alvéolaire correspondent à des aspects parfaitement définis sur le plan sémiologique avec une excellente concordance interobservateur. Certes l’imagerie paramétrique de la densité pulmonaire pourrait être utile sans et après injection de contraste en TDM ou en IRM pour évaluer la microperfusion du paren- chyme pulmonaire mais ces techniques sont du domaine de la recherche et le problème est particulièrement complexe en pathologie pulmonaire car la densité du poumon est tributaire de très nombreux paramètres polluants. Outre ce que vous avez mentionné il faut noter le volume pulmonaire d’acquisition qui n’est contrôlé que dans de rares études, les artefacts cinétiques cardiovasculaires, la complexité de la double circulation pulmonaire. Il y a des travaux sur le sujet dans la littérature et nous avons nous-mêmes tenté une étude de la microperfusion pulmonaire en IRM dans les fibroses mais le problème est bien plus complexe que pour le cerveau. Peut être y-a-t-il un avenir pour l’étude de la composante microperfusive de l’IRM de diffusion à b faible ? La surveillance TDM est généralement annuelle en dehors de tout épisode aigu ou de détérioration fonctionnelle rapide. Dans les évolutions progressives habituelles, les modifications TDM sont en retard de plusieurs mois sur les altérations fonctionnelles. Par contre l’aspect TDM du premier scanner est bon élément pronostic. Plus le rayon de miel est abondant et plus le pronostic est sombre.
M. Christian NEZELOF
L’histiocytose langerhansienne pose-t-elle un diagnostic différentiel ?
L’histiocytose langerhansienne ne pose pas de problème de diagnostic différentiel.
Certains auteurs ont décrit les aspects de kystes jointifs de l’histiocytose langerhansienne comme du rayon de miel mais c’est un abus de langage. A la différence des cavités en rayon de miel, les kystes jointifs de l’histiocytose langerhansienne sont plus volumineux, à paroi fine, de topographie aussi bien centrale que périphérique et surtout s‘accompagnent d’une augmentation du volume pulmonaire. Les kystes de l’histiocytose langerhansienne sont généralement très différents de ceux des autres maladies kystiques aussi bien dans l’évolution des lésions que dans leur aspect.
Bull. Acad. Natle Méd., 2010, 194, no 2, 353-365, séance du 2 février 2010