
Résumé
La lésion de la substantia nigra, au cours de la maladie de Parkinson, avait été soupçonnée dès la fin du 19e siècle par Brissaud, confirmée par Tretiakoff, mais négligée par Lewy. L’étude expérimentale du syndrome extrapyramidal provoqué par la réserpine conduisit Carlsson à découvrir le rôle de neuromédiateur joué par la dopamine, découverte qui aboutit rapidement au traitement par la L-DOPA. L’identification de la mutation du gène de l’alpha-synucléine chez quelques familles de maladie de Parkinson à transmission autosomique dominante fut suivie par la détection de la protéine dans les lésions. Elle est aujourd’hui reconnue comme le constituant principal des corps et des prolongements de Lewy. L’exceptionnelle sensibilité de l’immunohistochimie de l’alpha-synucléine contribua à mieux appréhender la diffusion des lésions dans ce qui fut appelé la maladie à corps de Lewy — un spectre pathologique qui couvre l’atteinte nerveuse périphérique celle du tronc cérébral dans la maladie de Parkinson, jusqu’aux lésions corticales de la démence à corps de Lewy. Le corps de Lewy lui-même ne semblent pas directement responsable des symptômes qui semble relever de la mort neuronale. La cause directe de la mort neuronale — anomalie métabolique provoquée par l’alpha-synucléine, saturation du système ubiquitineprotéasome, stress oxydatif — n’a pas encore été déterminée avec certitude. La mort cellulaire pourrait aussi être causée par des mécanismes extracellulaires comme l’inflammation ou la gliose.
Summary
Changes in the substantia nigra of patients with Parkinson’s disease were suspected by Brissaud in the late 19th century. They were subsequently confirmed by Tretiakoff but neglected by Lewy, who described the inclusion bodies that bear his name. The experimental Parkinsonian syndrome caused by reserpine led Carlsson to discover the neuromediatory role of dopamine, a finding at the origin of L-DOPA therapy. Identification of a mutation of the alpha-synuclein gene in cases of familial Parkinson’s disease with autosomal dominant transmission was followed by the detection of the protein product in Lewy bodies and neurites. Alpha-synuclein is now recognized as being the main constituent of Lewy bodies. Alpha-synuclein immunohistochemistry has revealed that lesions can extend from the autonomous nervous system to the cortex (in Lewy body dementia). The Lewy body itself does not appear to be the direct cause of symptoms, which correlate better with neuronal death. Neuronal death could be due to metabolic disturbances related to alpha-synuclein accumulation, ubiquitin-proteasome system dysfunction, or oxidative stress. Nonautonomous cell death, caused by neuro-inflammation or gliosis, has also been incriminated.
INTRODUCTION
Le processus pathologique de la maladie de Parkinson garde son secret depuis les premières descriptions anatomopathologiques datant de plus d’un siècle. Les progrès ont pourtant été importants faisant tour à tour appel à l’anatomie (topographie des lésions), la biochimie (découverte de la dopamine), la génétique (mutation de l’alpha-synucléine), la biologie cellulaire (rôle de l’ubiquitination) et aux modèles animaux — sujets que nous allons considérer successivement.
L’ANATOMIE
Le striatum et le pallidum ont longtemps été considérés comme le siège des lésions de la maladie de Parkinson. La substantia nigra, décrite par Vicq d’Azyr dans son traité d’anatomie (« J’appellerai cette espace tâche noire ou locus niger crurum cerebri ») [1], a été pour la première fois incriminée par Edouard Brissaud [2], à la suite d’un cas de syndrome parkinsonien asymétrique lié à une tumeur de la calotte mésencéphalique [3]. Dans son importante monographie ‘‘ Die Lehre vom Tonus und der Bewegung … ’’, Friedrich Lewy rechercha à l’échelon microscopique des stigmates de la maladie. Il décrivit des inclusions visibles dans les neurones et leurs prolongements après coloration par l’éosine. Pour Lewy, le tonus était réglé par deux centres antagonistes : un centre mésencéphalique dont la destruction provoquait une hypertonie, et un centre cérébelleux dont la destruction était associée à une hypotonie. Bien qu’il eut situé un hypothétique centre du tonus dans le mésencéphale, il ne considéra jamais qu’il pût s’agir de la substantia nigra. La raison en est probablement théorique : pour que Lewy puisse reconnaître le rôle joué par la substantia nigra, il aurait fallu qu’il en connût les connexions aux centres moteurs. À l’époque la cible des fins axones des neurones nigriques n’avait pas encore identifiée.
Tretiakoff [4], externe des hôpitaux, dans la clinique des maladies du système nerveux de Pierre Marie, portait aussi, curieusement, le titre de ‘‘ Chef du laboratoire de la Clinique des Maladies du Système Nerveux à La Salpêtrière ’’. Sa thèse démontrait que la substantia nigra était dépigmentée chez les patients parkinso- niens et que certains de ses neurones contenaient des corps d’inclusion sphériques auxquels il donna le nom de corps de Lewy. La perte neuronale observée dans la substantia nigra, ainsi que dans les autres noyaux pigmentés du tronc cérébral (locus coeruleus, noyau dorsal du vague) fut précisée par les travaux ultérieurs en particulier ceux de Hassler [5]. Les corps de Lewy sont encore utilisés aujourd’hui comme le marqueur pathologique de la maladie de Parkinson [6] et, du point de vue neuropathologique qui est celui des auteurs, sont une des clefs du mécanisme de la maladie. Ceci explique la place prépondérante qu’ils occupent dans cet exposé.
LA DOPAMINE
Le syndrome extrapyramidal des animaux d’expérience traités par la réserpine conduisit Arvid Carlsson à lier le trouble moteur à la déplétion dopaminergique du striatum et à démontrer que la dopamine jouait le rôle de neuromédiateur [7]. La mise en évidence des catécholamines par la méthode d’histofluorescence développée par Falck & Hillarp [8], également en Suède, permit de montrer que la substantia nigra était la source de la dopamine striatale. Elle était effondrée, chez l’homme, dans la maladie de Parkinson [9]. Ces découvertes successives conduisirent à un modèle réductionniste : la mort neuronale dans la substantia nigra conduit à l’appauvrissement dopaminergique du striatum, lui-même responsable du syndrome moteur. Ce paradigme est à l’origine de nombreux travaux, qui aboutirent, entre autres, à la mise au point du traitement par la L-DOPA [10, 11] et à l’apparition d’une maladie véritablement nouvelle du fait des répercussions profondes de cette nouvelle thérapeutique sur l’aspect « naturel » de l’affection [12].
L’ALPHA-SYNUCLÉINE
La génétique
Le caractère sporadique de la maladie de Parkinson idiopathique était une notion classique jusqu’à récemment. Le développement spectaculaire des outils de la génétique moléculaire conduisit à rechercher des cas familiaux. Quatre familles (une italienne et trois grecques), toutes originaires du pourtour méditerranéen furent ainsi identifiées. La transmission était autosomique dominante et le gène fut identifié de façon exceptionnellement rapide : il s’agissait du gène de l’alpha-synucléine (SNCA) [13]. Cette protéine avait été initialement isolée par Maroteaux et al. [14] au cours d’investigations visant à mettre en évidence de nouvelles protéines synaptiques. Le modèle utilisé était celui de l’organe électrique du poisson torpille, qui se comporte comme une synapse volumineuse. Le nom de la protéine témoigne de sa topographie synaptique et nucléaire (la localisation nucléaire n’a pas été retrouvée dans le tissu post-mortem humain). La protéine a été ‘‘ découverte ’’ au moins à deux autres reprises: sous le nom de synelfine dans une étude qui étudiait les protéines surexprimées au moment de l’apprentissage du chant chez le diamant mandarin (zebra finch) [15] et au cours d’un criblage systématique chez le porc (phosponeuroprotéine 14) [16]. Des anticorps anti-alpha synucléine avaient été obtenus au moment de la découverte de la protéine et il fut donc aisé de réaliser des études immunohistochimiques dès que la mutation de son gène fut mise en évidence.
Il apparut que tous les corps de Lewy étaient marqués par l’anticorps ; de nombreux prolongements dilatés, probablement axonaux, l’étaient aussi [17]. La sensibilité et la spécificité du marquage laissent penser que le constituant principal du corps de Lewy est en effet l’alpha-synucléine.
Les anticorps anti-alpha-synucléine constituaient un outil d’une exceptionnelle sensibilité pour réévaluer l’anatomie pathologique de la maladie. Il serait erroné de penser qu’il ait été indispensable à l’acquisition des notions nouvelles sur la maladie:
la présence de corps de Lewy dans le cortex cérébral avait été notée au moyen de la simple hématéine éosine dès 1961 [18] et Heiko Braak avait acquis une connaissance précise de la topographie élargie des lésions en utilisant une coloration argentique réputée spécifique (Campbell-Switzer method) [19]. Il n’en est pas moins vrai que l’immunohistochimie de l’alpha-synucléine permit d’analyser en routine des lésions jusqu’alors inconnues ou méconnues. Une nouvelle neuropathologie a ainsi vu le jour.
Les ‘‘ lésions de Lewy ’’
L’immunohistochimie révéla la fréquence de l’accumulation d’alpha-synucléine dans des structures situées en dehors des corps neuronaux. Il s’agit de prolongements dilatés, qui sont probablement plus nombreux que les corps d’inclusion eux-mêmes [20]. On appelait naguère ces accumulations des ‘‘ corps de Lewy extracellulaires ’’. Il est aujourd’hui établi qu’elles sont situées dans les prolongements neuronaux. Cette observation conduisit à forger le terme de prolongements (ou ‘‘ neurites ’’) de Lewy, corps et prolongements constituant la « pathologie de Lewy ». Il faut d’ailleurs signaler que ces lésions avaient été observées avec les moyens de l’époque et illustrées par Friedrich Lewy lui-même dans sa monographie ‘‘ Die Lehre vom Tonus und der Bewegung … ’’. Les corps et les prolongements sont habituellement associés mais il existe des dissociations : ainsi seuls des prolongements alpha-synucléine positifs sont habituellement constatés dans les secteurs CA2-3 de l’hippocampe [21], nous y reviendrons. L’immunohistochimie de l’alphasynucléine a permis non seulement de mieux analyser les accumulations cellulaires ;
elle a aussi conduit à mieux appréhender l’importante extension des lésions.
Les lésions extra-nigriques et les symptômes non-moteurs de la maladie de Parkinson.
La démence à corps de Lewy
La découverte de la diffusion de la pathologie de Lewy conduisit à dépasser le modèle réductionniste maladie de Parkinson = perte des neurones de la substantia nigra = manque de dopamine. La deuxième égalité reste exacte. Nous avons, par exemple, montré dans une étude clinico-pathologique prospective soutenue par une bourse de l’Académie nationale de médecine, qu’il existait en effet une excellente corrélation entre la perte des neurones nigriques et le syndrome extrapyramidal évalué par l’échelle motrice de l’UPDRS (Unified Parkinson Disease Rating Scale), un standard international d’évaluation du déficit extra-pyramidal [22]. Mais il serait erroné de réduire la maladie de Parkinson à son syndrome moteur comme on l’a souvent cru.
Des accumulations d’alpha-synucléine sont constatées dans d’autres structures du tronc cérébral: au niveau du bulbe elles siègent dans le noyau dorsal du vague et la zone réticulée intermédiaire qui l’entoure. Dans le pont, elles intéressent le complexe coeruleus-subcoeruleus. À l’étage sus-tentoriel, on les observe dans le bulbe olfactif, le noyau basal de Meynert et les noyaux du septum, l’amygdale temporale [23], les noyaux limbiques du thalamus [24], l’hypothalamus et le cortex cérébral. Des corps et des prolongements de Lewy sont aussi constatés dans le système nerveux végé- tatif : on en trouve dans les plexus de Meissner et d’Auerbach du tube digestif [25].
Ils sont aussi présents dans le ganglion stellaire et les plexus cardiaques : cette dernière localisation peut être détectée in vivo grâce à la scintigraphie au metaiodobenzylguanidine marqué sur l’iode 123 [26]. Tout récemment Heiko Braak et son équipe ont montré qu’ils étaient aussi présents dans les glandes salivaires, ouvrant de nouvelles perspectives diagnostiques [27].
Enfin, des corps de Lewy peuvent, dans certains cas, être mis en évidence dans l’hippocampe et le cortex cérébral. Dans une région limitée de l’hippocampe, à cheval sur les secteurs CA2 et CA3, des fibres synucléine positives sont révélées par les anticorps anti-alpha-synucléines. Ils peuvent être également présents dans le subiculum, le cortex entorhinal et transentorhinal, les cortex associatifs multimodaux et plus rarement uni-modaux.
La multiplicité des topographies lésionnelles soulève deux questions :
Témoigne-t-elle d’une progression chronologique de la pathologie ou au contraire d’atteintes relevant d’une sensibilité variable des tissus en réponse à une altération, métabolique ou d’autre nature, présente dans tout l’organisme ?
La première option est défendue par l’équipe d’Heiko Braak [28, 29] : selon cet auteur, la maladie de Parkinson débute dans les organes périphériques. Les lésions progressent ensuite le long des faisceaux nerveux en passant les synapses. L’atteinte du noyau dorsal du vague et de la zone réticulée intermédiaire dans le bulbe rachidien (stade 1) précède celle du complexe coeruleus-subcoeruleus dans le pont (stade 2), du mésencéphale, du noyau tubéromammillaire de l’hypothalamus et des noyaux magnocellulaires de la base du cerveau (stade 3), du néocortex temporal interne (stade 4), des néocortex associatifs pariétaux polymodaux (stade 5) et des cortex prémoteurs, primaires ou associatifs unimodaux (stade 6). Le bulbe olfactif et le noyau olfactif antérieur sont touchés précocement (dès le stade 1 ou 2) ; leur atteinte est sévère au stade 4. Ces lésions restent confinées aux structures olfactives et ne diffusent pas aux aires néocorticales voisines. L’amygdale temporale est intéressée dès le stade 4 et les lésions sont marquées aux stades ultérieurs. La pathologie de Lewy dans l’amygdale temporale est particulière : souvent intense, elle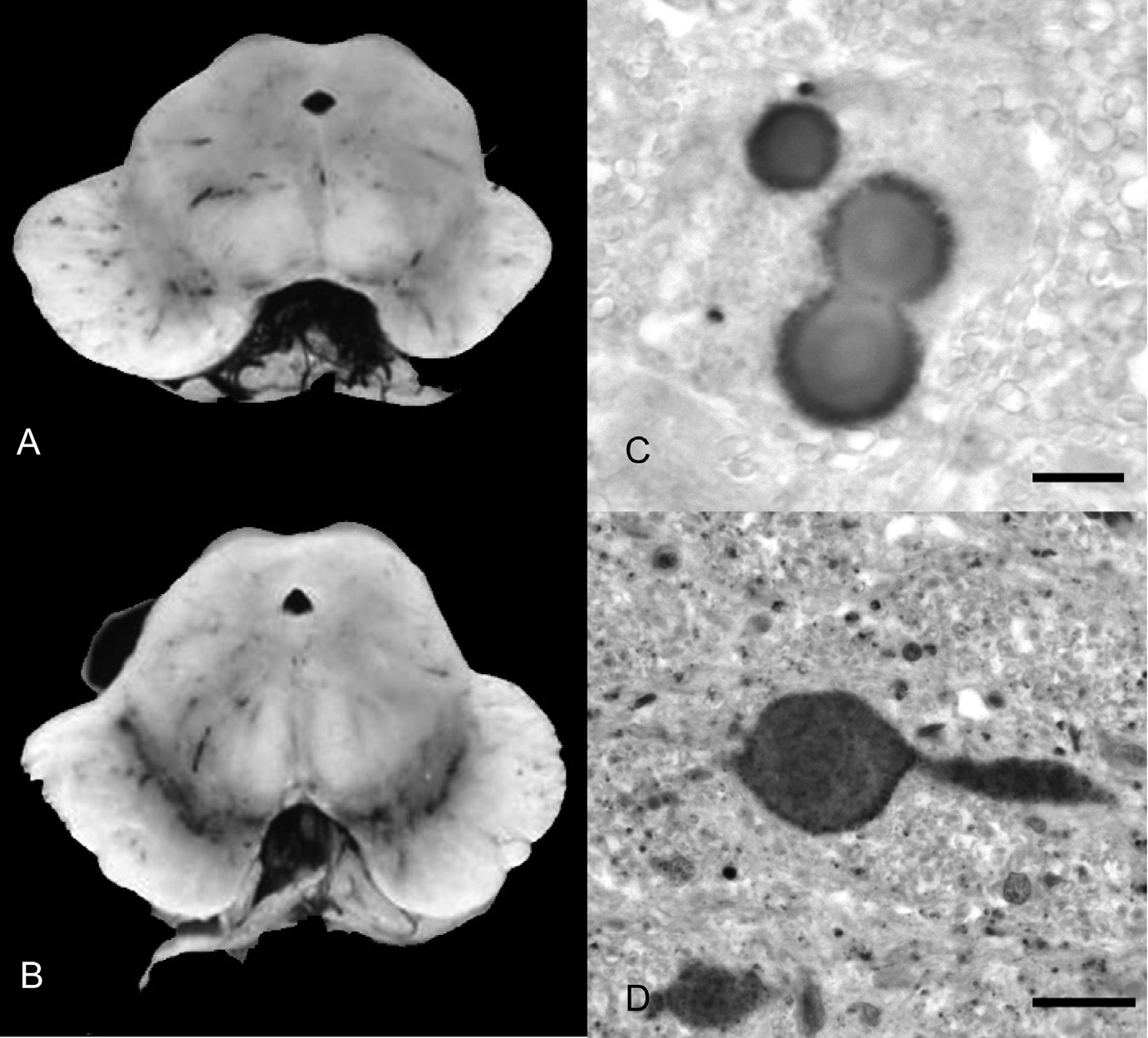
Fig. 1. — A et B : aspect macroscopique de la substantia nigra dépigmentée dans un cas de maladie de Parkinson (A) par rapport au témoin (B). Immunohistochimie de l’alpha-synucléine (révélation par la méthode biotine streptavidine peroxydase en utilisant la diaminobenzidine comme chromogène). L’anticorps marque trois corps de Lewy (colorés en marron) dans un neurone (C) et un prolongement axonal dilaté (D). Barre d’échelle = 5 microns.
est volontiers associée à la maladie d’Alzheimer. Dans ce dernier cas, des corps et des prolongements de Lewy peuvent n’être observés que dans l’amygdale temporale [30].
Braak et son équipe considèrent que cette progression provient de la transmission par voie nerveuse et synaptique d’un agent pathogène dont la nature n’est pas précisée [28]. Dans deux cas de greffe surrénale dans le striatum, des corps de Lewy se sont développés dans des cellules dopaminergiques fœtales transplantées [31, 32].
Ces observations exceptionnelles ont été interprétées comme la preuve d’une transmission synaptique de l’agent pathogène (quel qu’il soit) responsable de la formation des corps de Lewy. Un concept récent, initialement formulé au sujet du peptide Aβ de la maladie d’Alzheimer, fait jouer un rôle important aux agrégats protéiques encore solubles, qualifiés d’oligomères. C’est ainsi que l’oligomère d’alphasynucléine pourrait constituer l’espèce toxique [33]. Des données expérimentales laissent penser que l’agrégation de la protéine tau pourrait, dans la maladie d’Alzheimer, se transmettre d’un neurone à l’autre par l’intermédiaire des synapses.
Une hypothèse analogue peut être formulée pour la maladie de Parkinson [34].
Des critiques se sont élevées contre la conception d’une progression des lésions le long des connexions nerveuses à partir des plexus périphériques jusqu’au cortex en passant par le tronc cérébral et les noyaux sous-corticaux. Elle est en effet difficilement applicable aux cas génétiques liés à la mutation de l’alpha-synucléine : la mutation est évidemment présente dans toutes les cellules de l’organisme ; la topographie lésionnelle est mieux expliquée par des différences de sensibilité que par une progression synaptique d’un agent pathogène. D’autre part, plusieurs auteurs ont souligné des exceptions au schéma chronologique proposé par Braak [35, 36] : il n’est pas rare, par exemple, de ne trouver que des lésions minimes dans le tronc cérébral au cours des démences à corps de Lewy. Il a été remarqué que l’âge moyen des cas asymptomatiques aux stades 1 et 2 de la maladie est souvent plus élevé que celui des cas symptomatiques, une observation difficile à expliquer si l’on admet la progression chronologique proposée par Braak et al. , [35] du tronc cérébral au cortex. Enfin, même à un stade présymptomatique, deux évolutions des maladies à corps de Lewy semblent pouvoir être distinguées : débutant dans le tronc cérébral et poursuivant une progression ascendante comme dans le schéma de Braak et al. [29], ou d’emblée diffuse [37].
La multiplicité des sites lésionnels pourrait être expliquée, non par la progression des lésions, mais par des sensibilités différentes de certains neurones. Les ‘‘ maladies à corps de Lewy ’’ sont alors classées non par stades mais par ‘‘ types ’’ [38]. Kosaka et al. , reconnaissent trois types selon la topographie de la pathologie de Lewy: du tronc cérébral (dont le corrélat clinique est la maladie de Parkinson sans trouble cognitif), diffus où la pathologie concerne tant le cortex que le tronc cérébral (et dont le corrélat clinique est la démence à corps de Lewy ou la démence de la maladie de Parkinson), le type transitionnel enfin, comportant une pathologie du tronc cérébral associée à des lésions limitées au cortex limbique [38].
Existe-t-il une corrélation entre les lésions et les symptômes ?
La maladie de Parkinson a pu être considérée comme une affection purement motrice. La multiplicité des sites lésionnels est peu compatible avec cette opinion et laisse penser qu’une sémiologie clinique plus riche a été, à une époque, négligée à moins que la pathologie de Lewy ne provoque aucun déficit. Avec ou sans consé- quence fonctionnelle ? La question est d’importance car de la réponse qui y est apportée découle le rôle pathogénique qu’il faut attribuer aux corps et prolongements de Lewy et les orientations de la recherche thérapeutique qui en découle.
La conception selon laquelle la pathologie de Lewy n’est ni spécifique ni sensible a souvent été exprimée. Des points de vue extrêmes ont été soutenus : c’est ainsi que D. Calne [39] classe comme ‘‘ neuromythologique ’’ l’opinion selon laquelle le corps de Lewy a quelque chose à voir avec la maladie de Parkinson. Deux arguments principaux sont avancés : — des corps de Lewy peuvent être trouvés chez des patients asymptomatiques. Dans des grandes séries autopsiques (respectivement 904 et 1 241 cas) où ils ont été recherchés systématiquement, les corps de Lewy n’étaient symptomatiques que dans 20 à 30 % des cas [40, 41] ; — aucun corps de Lewy n’est mis en évidence dans d’authentiques maladies de Parkinson génétiques, comme celle liée à la mutation de la Parkine, de transmission autosomique récessive.
Le premier point ne soulève pas de réelles difficultés si l’on se souvient que le noyau dorsal du vague, la zone réticulée intermédiaire, le complexe coeruleussubcoeruleus, le bulbe olfactif sont atteints des années avant que la substantia nigra ne soit affectée à son tour : les symptômes provoqués par ces lésions passent le plus souvent inaperçus. Des corps de Lewy peuvent donc, en effet, être mis en évidence chez des patients qui ne souffrent pas de maladie de Parkinson et peuvent être considérés comme asymptomatiques. Si la diffusion des lésions augmentait de façon linéaire avec le temps, les chiffres rapportés de prévalence des corps de Lewy conduiraient à penser que la période asymptomatique de la maladie de Parkinson serait de trois à cinq fois plus longue que sa phase symptomatique (puisque seuls un tiers à un cinquième des cas autopsiques sont symptomatiques).
Le second point soulève la question de la définition de la maladie de Parkinson : la destruction de la substantia nigra, quelle que soit sa cause, provoque l’apparition du syndrome clinique moteur propre à l’affection : il est aussi bien constaté dans le tuberculome que dans la tumeur de la calotte mésencéphalique. La mutation de la parkine, portant sur les deux allèles, provoque une destruction de la substantia nigra : elle est, pour cette raison, responsable d’un syndrome parkinsonien, mais il pourrait bien s’agir d’une affection totalement différente de la maladie de Parkinson idiopathique, n’ayant en commun avec celle-ci que la mort des neurones de la substantia nigra. Et ce n’est pas parce que le gène porte le nom de parkine que sa mutation est à l’origine d’une maladie de Parkinson du même type que la forme idiopathique habituelle.
Il existe une dernière objection, plus difficile à lever, au rôle pathogène direct du corps de Lewy : il n’existe pas de corrélation (au sens de relation statistique entre deux quantités) entre le nombre de corps de Lewy par unité de volume (leur ‘‘ densité numérique ’’) et la sévérité des symptômes (appréciée par une échelle quantitative). Cette objection amène à distinguer deux façons d’aborder la question : selon la méthode classique des ‘‘ corrélations ’’ clinico-pathologiques, on peut déterminer si la présence de corps de Lewy est en général associée à un déficit fonctionnelle de la région intéressée. On peut aussi rechercher une liaison statistique entre la densité numérique des corps de Lewy et la sévérité des manifestations cliniques (corrélation au sens statistique).
Les corrélations clinico-pathologiques
La topographie lésionnelle correspond souvent à des symptômes cliniques identifiés. Lorsque ce n’est pas le cas, c’est le plus souvent en raison de la méconnaissance des conséquences cliniques du dysfonctionnement de la structure considérée. La corrélation clinico-pathologique la plus évidente concerne la substantia nigra. Les corps de Lewy y sont abondants et il est connu depuis l’observation de Brissaud que la destruction de la substantia nigra, quelle que soit sa cause, provoque un syndrome Parkinsonien. La présence de corps de Lewy dans le cortex cérébral est un marqueur à la fois sensible et spécifique des troubles cognitifs observés dans la maladie de Parkinson évoluée [42]. C’est aussi la caractéristique neuropathologique qui définit la démence à corps de Lewy. Il n’existe d’ailleurs aucune façon de distinguer ces deux affections (maladie de Parkinson avec démence et démence à corps de Lewy) sur les seuls critères neuropathologiques. L’interprétation des troubles cognitifs, initiaux ou associés à une maladie de Parkinson évoluée, est compliquée par la fréquente association aux lésions de Lewy, de plaques séniles et de dégénérescences neurofibrillaires, propres à la maladie d’Alzheimer. Cette question ne sera pas traitée ici faute de place. Nous nous contenterons de signaler que les nouveaux critères clinico-pathologiques des démences à corps de Lewy consignent les lésions de Lewy et celles d’Alzheimer afin de comparer leur intensité [43].
Des corps de Lewy sont aussi présents dans le bulbe olfactif : l’anosmie peut précéder le syndrome Parkinsonien de plusieurs années [44]. L’un des exemples les plus frappants des corrélations clinico-pathologiques récentes rendues possibles par les nouvelles observations neuropathologiques est celui des troubles du comportement lors du sommeil paradoxal (RBD : Rapid eye movement sleep Behavior Disorder). Il s’agit de rêves vécus avec une intensité particulière, souvent effrayants, associés à des comportements moteurs simples ou complexes parfois violents.
Comme tous les rêves, ces épisodes surviennent lors du sommeil paradoxal. Une proportion élevée de ces cas développent dans les années, parfois les décennies qui suivent, les signes moteurs de la maladie de Parkinson. Le RBD est présent dans 33 à 60 % des cas de maladie de Parkinson, et, dans 50 à 80 % des cas de démence à corps de Lewy [45]. Les connaissances actuelles sur le sommeil paradoxal indiquent que ces symptômes sont probablement liés à l’atteinte du complexe coeruleussubcoeruleus dans le pont. En prenant le syndrome RBD comme un marqueur clinique de cette lésion, on peut en déduire que l’atteinte du pont précède celle du mésencéphale de plusieurs années. La pathologie de Lewy de siège périphérique a aussi fait l’objet d’études : la constipation par exemple pourrait être la conséquence des lésions des plexus de Meissner et d’Auerbach. De fait, une constipation sévère est plus fréquente dans une population de patients qui ont développé des années plus tard un syndrome Parkinsonien que dans le groupe de contrôle. C’est aussi le cas pour la dysautonomie.
Les conséquences cliniques des lésions de l’amygdale temporale, des secteurs CA2-3 de l’hippocampe, des noyaux thalamiques ou hypothalamiques sont mal connues.
Les hallucinations visuelles sont statistiquement liées à la densité des corps de Lewy dans l’amygdale temporale, la circonvolution para-hippocampique, et le cortex temporal inférieur [46]. Elles constituent un signe très spécifique des maladies à corps de Lewy [47].
Le rapport entre la densité numérique des corps de Lewy et l’intensité des symptômes
Au cours d’une étude mentionnée plus haut, nous avions pu constater que le syndrome extrapyramidal (évalué par l’échelle motrice de l’UPDRS) était lié de façon statistique à la mort neuronale dans la substantia nigra. La droite de corré- lation indiquait que la perte d’un point sur l’échelle motrice équivalait en moyenne à la perte de 33 neurones/mm3 dans la substantia nigra. D’autre part, nous avions montré (comme d’autres l’avaient fait au moyen de marqueurs radioactifs in vivo ) que la perte neuronale au cours du temps suivait une courbe exponentielle négative :
la mort neuronale la plus sévère était observée durant les premières années (la substantia nigra perd environ la moitié de ses neurones au cours des cinq premières années de la maladie). L’extrapolation de la courbe suggérait une phase pré- diagnostique de quatre ans et demi. Cependant, aucune corrélation n’a pu être trouvée entre la densité numérique des corps de Lewy et l’intensité du déficit moteur ou l’ampleur de la mort neuronale. Dans une étude ultérieure [48], nous avons observé que la densité numérique des corps de Lewy restait remarquablement constante: quatre pour cent des neurones de la substantia nigra, en moyenne, sont porteurs de corps de Lewy que la maladie soit peu avancée et les neurones encore nombreux ou, au contraire, qu’il s’agisse d’un stade terminal où seuls quelques neurones subsistent. Le nombre total de corps de Lewy présents dans la substantia nigra à un moment donné dépend donc du nombre de neurones survivants : il paraîtra élevé au début de la maladie (quatre pour cent d’environ mille neurones présents sur une coupe) ; il sera au contraire très faible à un stade avancé (quelques pour cent d’une centaine de neurones). Ceci explique pourquoi dans certains cas de nombreux corps de Lewy sont observés alors que les symptômes sont absents ou discrets : au début de l’affection, les corps de Lewy paraissent nombreux mais ils sont en fait trop rares (ils n’intéressent toujours que 4 % des neurones) pour provoquer des symptômes. C’est lorsque la mort neuronale a éliminé un nombre suffisant de neurones que les premiers symptômes apparaissent. Il est habituel de dire qu’une perte des deux tiers des neurones est nécessaire avant l’apparition des premiers symptômes. L’origine de cette valeur classique est obscure et ne paraît pas assise sur des données quantitatives sérieuses. Dans notre étude, le diagnostic de maladie de Parkinson avait été porté chez des patients qui avaient perdu ‘‘ seulement ’’ 20 % de la population neuronale de la substantia nigra.
Il s’agit maintenant de comprendre comment le pourcentage de neurones comportant des corps de Lewy reste en moyenne constant au cours du temps. Si les corps de Lewy n’étaient pas résorbés, leur nombre ne devrait cesser d’augmenter (c’est ce qui se produit, par exemple, pour les dégénérescences neurofibrillaires de la maladie d’Alzheimer). Comme ce n’est pas le cas, il doit exister un équilibre entre la production des corps de Lewy et leur résorption. Le modèle le plus simple permettant d’expliquer cet équilibre est celui qui suppose la mort des neurones porteurs de corps de Lewy, eux-mêmes éliminés par les macrophages dans le milieu extracellulaire. Dans ce modèle, la durée de vie d’un neurone porteur d’un corps de Lewy est d’environ six mois [48].
Des modèles alternatifs ont été développés. L’opinion est aujourd’hui largement répandue que le corps de Lewy n’est pas toxique mais protecteur [49]. Le terme est ambigu : il peut signifier que parmi les neurones de la substantia nigra, ceux qui ne comportent pas de corps de Lewy meurent plus vite. S’il en était ainsi, la proportion de neurones porteurs de corps de Lewy ne devrait cesser d’augmenter au cours de la maladie, contrairement à ce qui est observé. Le terme de protecteur peut aussi être utilisé pour indiquer que la maladie de Parkinson évoluerait beaucoup plus vite si, dans le processus lésionnel général, il n’y avait pas formation de corps de Lewy. C’est ce que montrent les modèles cellulaires : l’agrégation de l’alpha-synucléine évite que des espèces toxiques diffusent librement dans la cellule [49]. La réalité de ce mécanisme chez l’homme in vivo doit encore être assurée.
LES MÉCANISMES DE LA MORT NEURONALE
La pathologie de Lewy est, par définition, liée à la maladie de Parkinson, mais le mécanisme de sa formation et la cause de la mort neuronale qui accompagne sa présence, restent mal compris.
Le corps de Lewy et le système ubiquitine-protéasome (UPS)
L’ubiquitine (dont la découverte valut à A. Ciechanover, A. Hershko et I. Rose le prix Nobel 2004) est une petite protéine qui se fixe sur les résidus lysine des protéines mal repliées. Les molécules ainsi marquées par une courte chaîne de polyubiquitine sont transportées et détruites par le protéasome. L’immunohistochimie antiubiquitine sur des coupes de tissu permet de mettre en évidence les protéines mal repliées. Ces anticorps se fixent sur les corps et les prolongements de Lewy qui les accompagnent [50] ; la présence d’ubiquitine témoigne de la présence de protéines mal repliées au sein des lésions. Il est évidemment tentant de penser que l’alphasynucléine est la protéine mal repliée à laquelle est fixée la chaîne de polyubiquitine mais c’est en réalité la synphiline-1, une protéine associée à l’alpha-synucléine [51].
La présence d’ubiquitine dans les corps et les prolongements de Lewy ont laissé penser qu’une déficience du système ubiquitine protéasome pouvait être au cœur des mécanismes pathologiques. De fait l’inhibition du protéasome par injection systé- mique d’inhibiteur provoque, chez le rat, l’apparition d’une accumulation d’alphasynucléine et une mort neuronale [52], résultat contesté dans la littérature car reproduit seulement par un petit nombre d’équipes. Cependant, l’inactivation du constituant 26s de protéasome par manipulation génétique [53] semble provoquer les même lésions et tend à démontrer que le dysfonctionnement du système UPS est aujourd’hui l’un des mécanismes explicatifs potentiels les plus prometteurs.
Le stress oxydatif et la mitochondrie [54]
L’activité, non contrôlée par la cellule, d’espèces réactives de l’oxygène (ROS) dans la substantia nigra est attestée par de nombreux indices dans la maladie de Parkin- son comme dans le modèle MPTP : la concentration d’acides gras polyinsaturés est diminuée, la concentration de 4-hydroxy-2 nonénal, un produit de peroxydation de l’acide arachidonique, abondant dans les membranes, est au contraire augmentée.
Les molécules d’hydroxynonénal réagissent avec l’alpha-synucléine dont elles pourraient entraver la fonction physiologique.
Les espèces réactives de l’oxygène pourraient être issues de la mitochondrie (elles sont produites à la suite d’une inhibition du complexe 1, par exemple dans le modèle MPTP ou roténone). Selon une autre hypothèse, la dopamine quitterait les vésicules synaptiques (légèrement acides) pour le cytoplasme, où elle serait auto-oxydée et produirait les espèces réactives. Il faut cependant noter, à l’encontre de ce mécanisme supposé, que les animaux dépourvus, par manipulation génétique, de tyrosine hydroxylase (donc incapables de produire de la dopamine), sont aussi sensibles au MPTP. Les espèces réactives de l’oxygène peuvent aussi être produites par les cellules gliales ou inflammatoires.
Les causes extraneuronales de la mort cellulaire : l’inflammation
De nombreux travaux récents ont tenté de déterminer si l’inflammation et la réaction gliale, naguère considérées comme la conséquence de la mort neuronale, étaient susceptibles, au contraire, de la perpétuer par une action toxique des médiateurs libérés [55]. La mort neuronale, lorsqu’elle est provoquée par des causes extérieures à la cellule, a été appelée ‘‘ non-autonome ’’ [56]. C’est ainsi par exemple que la destruction cellulaire se poursuit des années après l’exposition au MPTP [57].
Des signes d’activation de la microglie (l’induction de la synthétase du monoxyde d’azote ou iNOS) et d’inflammation sont présents dans la substantia nigra [58].
Stress oxydatif ou inflammation, il est aujourd’hui très difficile de déterminer s’il s’agit de cause ou de conséquence de la mort neuronale.
D’AUTRES MUTATIONS, DE NOUVELLES QUESTIONS
L’alpha-synucléine joue un rôle central dans la pathologie, non seulement parce que la mutation de son gène est associée à une maladie de Parkinson ou à une démence à corps de Lewy de transmission autosomique dominante, mais aussi parce qu’elle paraît être le constituant principal des lésions. D’autres mutations ont été associées à des maladies de Parkinson autosomiques dominantes ou récessives [59]. Il est encore trop tôt pour déterminer si les protéines intéressées sont toutes impliquées dans une même voie métabolique et, faute de place, nous ne considérerons que quelques points particuliers.
Les formes autosomiques dominantes (mutations du gène de l’alpha-synucléine ou de la kinase 2 à répétitions riches en leucine =LRRK2) sont associées à une pathologie de Lewy typique, souvent grave et responsable de démence [60]. La pathologie des formes récessives en particulier de la mutation de la Parkine est en général dépourvue de corps de Lewy et n’est pas associée à un déficit cognitif. La parkine a une activité de type E3 ligase, une des étapes de l’activation de l’ubiquitine.
Un cas particulièrement intéressant est celui de la mutation de la glucocérébrosidase. La mutation est, à l’état homozygote, associée à la maladie de Gaucher. A l’état hétérozygote, elle joue le rôle de facteur de risque des maladies à corps de Lewy (maladie de Parkinson et démence à corps de Lewy). Il est possible qu’ici encore, ce soit le système ubiquitine-protéasome qui soit en cause : la protéine mutée, dégradée par ce système, pourrait le saturer.
Ces quelques exemples montrent que plusieurs des mutations identifiées intéressent des protéines impliquées dans le système ubiquitine-protéasome mais il est impossible aujourd’hui de déterminer si les mécanismes de la mort neuronale sont les mêmes dans la maladie de Parkinson idiopathique et dans les différentes formes génétiques.
CONCLUSIONS
La définition anatomo-pathologique de la maladie de Parkinson comporte deux éléments : la mort neuronale dans des régions spécifiques du cerveau et la présence d’une pathologie de Lewy. Quel est le mécanisme de l’accumulation de l’alphasynucléine dans les corps et prolongements de Lewy ? Quelle est la conséquence de cette accumulation ? Provoque-t-elle un dysfonctionnement cellulaire ? Est-elle la cause de la mort neuronale ? Les progrès de la biologie moléculaire ont permis d’identifier les protéines en cause mais l’enchaînement des évènements est encore mal compris. La compréhension de cette cascade de réactions pathologiques constitue la prochaine étape de l’effort de recherche entrepris depuis plus d’un siècle pour élucider la cause de la maladie et la traiter.
REMERCIEMENTS
L’auteur tient à remercier l’équipe technique du laboratoire de Neuropathologie Raymond Escourolle pour la qualité de son travail.
BIBLIOGRAPHIE [1] Vicq D’Azyr F. — Traité d’anatomie et de physiologie avec des planches coloriées représentant au naturel les divers organes de l’homme et des animaux Series in Traité d’anatomie et de physiologie avec des planches coloriées représentant au naturel les divers organes de l’homme et des animaux. Didot, Paris, 1 , 1786.
[2] Brissaud E. — Nature et pathogénie de la maladie de Parkinson., in Leçons sur les maladies nerveuses. Masson, Paris. p. 488-501, 1895.
[3] Blocq P., Marinesco G. — Sur un cas de tremblement parkinsonien hémiplégique, symptomatique d’une tumeur du pédoncule cérébral . CR Soc. Biol., Paris , 1893, 5 , 105-111.
[4] Lees A.J., Selikhova M., Andrade L.A., Duyckaerts C. — The black stuff and Konstantin Nikolaevich Tretiakoff . Mov. Disord. , 2008, 23 , 777-83.
[5] Hassler R. — Zur Pathologie der Paralysis agitans und des postenzephalitischen Parkinsonismus. J. Psychol. Neurol. , 1938, 48 , 387-476.
[6] Dickson D.W., Braak H., Duda J.E., Duyckarts C., Gasser T., Halliday G.M. et al. —
Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria . Lancet Neurol. , 2009, 8 , 1150-7.
[7] Carlsson A. — A half-century of neurotransmitter research: impact on neurology and psychiatry. Nobel lecture . Biosci. Rep. , 2001, 21 , 691-710.
[8] Falck B., Hillarp N.A., Thieme G., Torp A. — Fluorescence of catecholamine and related compounds condensed with formaldehyde. J. Histochem Cytochem , 1962, 10 , 348-354.
[9] Ehringer H., Homykiewicz.
— Verteilung von Noradrenalin und Dopamin (3-Hydroxytyramin) im Gehirn des Menschen und ihr Verhalten bei Erkrankungen des Extrapyramidalen Systems. Klin Wochenschr , 1960, 38 , 1236-1239.
[10] Cotzias G.C., Van Woert M.H., Schiffer L.M. — Aromatic amino acids and modification of parkinsonism . N. Engl. J. Med. , 1967, 276 , 374-9.
[11] Boudin G., Castaigne P., Lhermitte F., Beck H., Guillard A., Marteau R. et al. —
Traitement de la maladie de Parkinson par la L-Dopa. Soixante-dix-sept cas.
Rev. Neurol., (Paris) , 1970, 122 , 89-102.
[12] Cambier J. — Le Parkinson traité par la L. Dopa, les trente ans d’une maladie nouvelle . Presse
Med. , 1997, 26 , 1347-9.
[13] Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A. et al. —
Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease.
Science , 1997, 276 , 2045-2047.
[14] Maroteaux L., Campanelli J.T., Scheller R.H. — Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. , 1988, 8 , 2804-2815.
[15] George J.M., Jin H., Woods W.S., Clayton D.F. — Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. , 1995, 15 , 361-372.
[16] Jakes R., Spillantini M.G., Goedert M. — Identification of two distinct synucleins from human brain . FEBS Lett. , 1994, 345 , 27-32.
[17] Spillantini M.G., Schmidt M.L., Lee V.M.Y., Trojanowski J.Q., Jakes R., Goedert M. — Alpha-synuclein in Lewy bodies . Nature , 1997, 388 , 839-840.
[18] Okazaki H., Lipkin L.E., Aronson S.M. — Diffuse intracytoplasmic inclusions (Lewy type) associated with progressive dementia and quadriparesis in flexion. J. Neuropathol. Exp. Neurol. , 1961, 20 , 237-244.
[19] Braak H., Braak E., Yilmazer D., Schultz C., Devos R.A.I., Jansen E.N.H. — Nigral and extranigral pathology in Parkinson’s disease . J Neural Transm , 1995, Supp 46 , 15-31.
[20] Braak H., Sandmann-Keil D., Gai W., Braak E. — Extensive axonal Lewy neurites in Parkinson’s disease: a novel pathological feature revealed by alpha-synuclein immunocytochemistry. Neurosci. Lett. , 1999, 265 , 67-69.
[21] Dickson D.W., Schmidt M.L., Lee V.M.Y., Zhao M.L., Yen S.H., Trojanowski J.Q. — Immunoreactive profile of hippocampal Ca2/3 neurites in diffuse Lewy body disease. Acta Neuropathol (Berl) , 1994, 87 , 269-276.
[22] Greffard S., Verny M., Bonnet A.M., Beinis J.Y., Gallinari C., Meaume S. et al. — Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy bodyassociated neuronal loss in the substantia nigra . Arch. Neurol. , 2006, 63 , 584-8.
[23] Braak H., Braak E., Yilmazer D., Devos R.A.I., Jansen E.N.H., Bohl J. , et al. — Amygdala pathology in Parkinson’s disease . Acta Neuropathol (Berl) , 1994, 493-500.
[24] Rub U., Del Tredici K., Schultz C., Ghebremedhin E., De Vos R.A., Jansen Steur E. , et al.
— Parkinson’s disease: the thalamic components of the limbic loop are severely impaired by alpha-synuclein immunopositive inclusion body pathology . Neurobiol. Aging , 2002, 23 , 245-54.
[25] Lebouvier T., Chaumette T., Damier P., Coron E., Touchefeu Y., Vrignaud S. et al. —
Pathological lesions in colonic biopsies during Parkinson’s disease . Gut , 2008, 57 , 1741-3.
[26] Yoshita M. — Differentiation of idiopathic Parkinson’s disease from striatonigral degeneration and progressive supranuclear palsy using iodine-123 meta-iodobenzylguanidine myocardial scintigraphy . J. Neurol. Sci. , 1998, 155 , 60-7.
[27] Del Tredici K., Hawkes C.H., Ghebremedhin E., Braak H. — Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease . Acta Neuropathol. , 119 , 703-13.
[28] Del Tredici K., Rub U., De Vos R.A., Bohl J.R., Braak H. — Where does parkinson disease pathology begin in the brain? J. Neuropathol. Exp. Neurol. , 2002, 61 , 413-426.
[29] Braak H., Del Tredici K., Rub U., De Vos R.A., Jansen Steur E.N., Braak E. — Staging of brain pathology related to sporadic Parkinson’s disease . Neurobiol. Aging , 2003, 24 , 197-211.
[30] Uchikado H., Lin W.L., Delucia M.W., Dickson D.W. — Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy . J. Neuropathol. Exp. Neurol. , 2006, 65 , 685-97.
[31] Kordower J.H., Chu Y., Hauser R.A., Freeman T.B., Olanow C.W. — Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease . Nat. Med , 2008, 14 , 504-6.
[32] Li J.Y., Englund E., Holton J.L., Soulet D., Hagell P., Lees A.J. et al. — Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation .
Nat. Med. , 2008, 14 , 501-3.
[33] Brown D.R. — Oligomeric alpha-synuclein and its role in neuronal death . IUBMB Life , 62 , 334-9.
[34] Clavaguera F., Bolmont T., Crowther R.A., Abramowski D., Frank S., Probst A. et al. —
Transmission and spreading of tauopathy in transgenic mouse brain . Nat. Cell Biol. , 2009, 11 , 909-13.
[35] Halliday G.M., Del Tredici K., Braak H. — Critical appraisal of brain pathology staging related to presymptomatic and symptomatic cases of sporadic Parkinson’s disease. J. Neural.
Transm. , 2006, Suppl. 70 , 99-103.
[36] Kalaitzakis M.E., Graeber M.B., Gentleman S.M., Pearce R.K. — The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: a critical analysis of alpha-synuclein staging . Neuropathol. Appl. Neurobiol. , 2008, 34 , 284-95.
[37] Frigerio R., Fujishiro H., Ahn T.B., Josephs K.A., Maraganore D.M., Delledonne A. et al. — Incidental Lewy body disease: Do some cases represent a preclinical stage of dementia with Lewy bodies?
Neurobiol. Aging , 2009.
[38] Kosaka K., Tsuchiya K., Yoshimura M. — Lewy body disease with and without dementia: a clinicopathological study of 35 cases. Clin. Neuropathol. , 1988, 7 , 299-305.
[39] Calne D.B., Mizuno Y. — The neuromythology of Parkinson’s Disease . Parkinsonism Relat
Disord , 2004, 10 , 319-22.
[40] Parkkinen L., Kauppinen T., Pirttila T., Autere J.M., Alafuzoff I. — Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia . Ann. Neurol. , 2005, 57 , 82-91.
[41] Saito Y., Ruberu N.N., Sawabe M., Arai T., Kazama H., Hosoi T. et al. — Lewy bodyrelated alpha-synucleinopathy in aging.
J. Neuropathol. Exp. Neurol. , 2004, 63 , 742-749.
[42] Hurtig H.I., Trojanowski J.Q., Galvin J., Ewbank D., Schmidt M.L., Lee V.M. et al. —
Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease . Neurology , 2000, 54 , 1916-21.
[43] McKeith I.G., Dickson D.W., Lowe J., Emre M., O’Brien J.T., Feldman H. , et al. —
Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium . Neurology , 2005, 65 , 1863-72.
[44] Hawkes C.H., Shephard B.C., Daniel S.E. — Olfactory dysfunction in Parkinson’s disease.
J.
Neurol. Neurosurg. Psychiatry , 1997, 62 , 436-446.
[45] Boeve B.F., Silber M.H., Saper C.B., Ferman T.J., Dickson D.W., Parisi J.E. , et al. —
Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease .
Brain , 2007, 130 , 2770-88.
[46] Harding A.J., Broe G.A., Halliday G.M. — Visual hallucinations in Lewy body disease relate to Lewy bodies in the temporal lobe . Brain , 2002, 125 , 391-403.
[47] Williams D.R., Lees A.J. — Visual hallucinations in the diagnosis of idiopathic Parkinson’s disease: a retrospective autopsy study . Lancet Neurol. , 2005, 4 , 605-10.
[48] Greffard S., Verny M., Bonnet A.M., Seilhean D., Hauw J.J., Duyckaerts C. — A stable proportion of Lewy body bearing neurons in the substantia nigra suggests a model in which the Lewy body causes neuronal death . Neurobiol. Aging , 2008, 1558-1497.
[49] Tanaka M., Kim Y.M., Lee G., Junn E., Iwatsubo T., Mouradian M.M. — Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective . J. Biol. Chem. , 2004, 279 , 4625-31.
[50] Lowe J., Blanchard A., Morrell K., Lennox G., Reynolds L., Billett M. et al. —
Ubiquitin is common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and Mallory bodies in alcoholic liver disease . J. Pathol. , 1988, 155 , 9-15.
[51] Lim K.L., Chew K.C., Tan J.M., Wang C., Chung K.K., Zhang Y. , et al. — Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation . J. Neurosci. , 2005, 25 , 2002-9.
[52] McNaught K.S., Olanow C.W. — Proteasome inhibitor-induced model of Parkinson’s disease . Ann. Neurol. , 2006, 60 , 243-7.
[53] Bedford L., Hay D., Devoy A., Paine S., Powe D.G., Seth R. et al. — Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies . J. Neurosci. , 2008, 28 , 8189-98.
[54] Zhou C., Huang Y., Przedborski S. — Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance . Ann. N Y Acad. Sci. , 2008, 1147 , 93-104.
[55] Hirsch E.C., Breidert T., Rousselet E., Hunot S., Hartmann A., Michel P.P. — The role of glial reaction and inflammation in Parkinson’s disease . Ann. N Y Acad. Sci. , 2003, 991 , 214-28.
[56] Dawson T.M. — Non-autonomous cell death in Parkinson’s disease . Lancet Neurol. , 2008, 7 , 474-5.
[57] Langston J.W., Forno L.S., Tetrud J., Reeves A.G., Kaplan J.A., Karluk D. — Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine exposure . Ann. Neurol. , 1999, 46 , 598-605.
[58] Hirsch E.C., Hunot S. — Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. , 2009, 8 , 382-97.
[59] Lesage S., Brice A. — Parkinson’s disease: from monogenic forms to genetic susceptibility factors . Hum. Mol. Genet. , 2009, 18 , R48-59.
[60] Vitte J., Traver S., Maues De Paula A., Lesage S., Rovelli G., Corti O. et al. —
Leucine-Rich Repeat Kinase 2 is associated with the endoplasmic reticulum in dopaminergic neurons and accumulates in the core of Lewy bodies in Parkinson disease . J. Neuropathol. Exp.
Neurol ., 2010, 69 , 959-972.
DISCUSSION
M. Bernard LECHEVALIER
Quels sont les rapports : (différence et similitude) de la dopamine de l’aire tegmentale ventrale et du locus Niger ? Qu’en est-il de l’aire tegmentale ventrale dans la maladie de Parkinson évoluée ?
L’aire tegmentale ventrale est un sous-noyau, situé à la partie interne et dans la bande dorsale (‘dorsal tier’) de la substantia nigra. Elle est dopaminergique et sa dopamine est la même que celle qui est produite par les autres sous-noyaux. Elle se différencie par ses connexions corticolimbiques et avec le nucleus accumbens, lui-même structure centrale du système de récompense. Ce sont ces dernières connexions qui laissent penser que l’aire tegmentale ventrale est impliquée dans les troubles du comportement, notamment le jeu pathologique, traités par M. Césaro au cours de cette séance.
M. François-Bernard MICHEL
Quelle fut la participation de Brissaud dans les premières contributions à la connaissance de la substantia Nigra ?
Brissaud décrivit un syndrome hémi-parkinsonien associé à un processus expansif localisé à la substantia nigra contralatérale. C’est, à partir de cette observation concrète qu’il suggéra la relation entre substantia nigra et maladie de Parkinson.
Mme Monique ADOLPHE
Avez-vous été conduit à une hypothèse de traitement en partant de l’alpha-synucléine ?
La surexpression de l’alpha-synucléine chez la souris transgénique ne provoque pas l’apparition de lésions semblables à celles qui sont observées dans la maladie de Parkinson — en particulier, la pathologie de type Lewy fait défaut. En revanche, l’inhibition du système ubiquitine-protéasome est associée à la présence de corps d’inclusion, ressemblant au corps de Lewy. L’hypothèse a été avancée que la surcharge de ce système était responsable de la pathologie, mais, à ma connaissance, elle n’a encore débouché sur aucune piste thérapeutique sérieuse.
M. Alim-Louis BENABID
Au récent congrès WPC à Glasgow, une hypothèse a été signalée selon laquelle la progression du processus neuro-dégénératif pourrait découler d’une diffusion de l’alpha-synucléine qui jouerait un rôle « contaminant » similaire ou comparable à celui des prions.
L’hypothèse de la transmission de la pathologie par les connexions nerveuses (comme se propagent les lésions provoquées par la PrPsc) repose principalement sur deux types de données : — L’examen post mortem a révélé que des cellules greffées dans le striatum pour corriger le déficit dopaminergique développaient, des années après l’intervention, une pathologie de type Lewy. L’explication la plus communément admise est celle d’une transmission d’un agent pathogène hypothétique par les connexions qui s’établissent entre le tissu hôte et le greffon. — Heiko Braak et coll. , ont montré qu’il existait des stades de développement de la pathologie de type Lewy, touchant d’abord la zone réticulée intermédiaire du bulbe, puis le locus coeruleus dans le pont, la substantia nigra dans le mésencéphale, le noyau basal de Meynert et les cortex limbiques, enfin le néocortex. Il est possible que l’agent pathogène hypothétique se propage le long des connexions qui relient ces différentes structures. L’origine entérique de l’agent pathogène a même été suggérée du fait de la présence de lésions dans les plexus nerveuw intestinaux, lésions étudiées par exemple par l’équipe de Pascal Derkinderen.
* Laboratoire de neuropathologie Escourolle — Hôpital de la Pitié Salpêtrière — 47 bld de l’Hôpital, 75651 Paris cedex 13 ; e-mail : charles.duyckaerts@psl.aphp.fr Tirés à part : Professeur Charles Duyckaerts, même adresse Article reçu le 6 septembre 2010, accepté le 4 octobre 2010
Bull. Acad. Natle Méd., 2010, 194, no 7, 1287-1304, séance du 12 octobre 2010