
Résumé
Diverses anomalies cérébrales concomitantes des troubles schizophréniques sont aujourd’hui identifiées : signes neurologiques certes mineurs et peu spécifiques, anomalies cognitives et neurophysiologiques. En outre, les données de l’imagerie cérébrale ont permis d’observer des anomalies précoces telles des anomalies de la gyration. Ces particularités sont également retrouvées chez des apparentés indemnes de symptômes schizophréniques. Ceci pose la question des déterminants d’un passage de l’état de vulnérabilité à celui de trouble avéré. Les études de biologie moléculaire ont mis en évidence des anomalies dans l’expression de facteurs impliqués dans la plasticité cellulaire ou au niveau de protéines structurales : reeline, GAD, BDNF, gène du D3-R. Tous ces résultats ont réorienté les études du génome difficiles à répliquer à partir de phénotypes aussi hétérogènes que la maladie elle-même. Les recherches tentent désormais d’identifier des caractéristiques phé- notypiques dont le déterminisme génétique est plus simple que celui de la maladie : modulation de l’onde P50, gène de la COMT, du BDNF ou d’autres protéines. La question posée étant désormais celle de l’identification d’indices de vulnérabilité et des mécanismes de transition pour accéder à une prévention efficace.
Summary
Brain anomalies associated with schizophrenic disorders may be of a cognitive, neurophysiological or neurological nature [the latter being relatively minor and nonspecific]. Brain imaging has revealed early anomalies such as cortical-subcortical atrophy and abnormal gyration. These anomalies can also be present in relatives free of schizophrenic symptoms. This raises the question of what determines the transition from vulnerability to clinical onset. There is now evidence that schizophrenic disorders are true brain diseases. This is based on neuropathological studies, brain imaging and clinical findings such as ‘‘ soft ’’ neurological signs (pyramidal and extrapyramidal symptoms, coordination difficulties, etc.). Cognitive dysfunctions such as attention and memory disorders and abnormal verbal fluency have also been described. Oculomotor pursuit and auditive evoked potentials have identified specific neurophysiological disorders such as N300 and P50 wave modifications. Schizophrenic disorders can also be associated with neuronal abnormalities, notably affecting factors involved in synaptic transmission and plasticity. For example, BDNF protein deficit is linked to certain late-onset forms of schizophrenia. Genetic studies are no longer focusing on a possible disease genotype but rather on phenotypic characteristics determined by simpler genotypes (P50 wave modulation, COMT and BDNF genes). The ultimate objective is to identify high-risk subjects, in order to shorten the treatment delay and thereby improve long-term outcome. The benefit of primary prophylaxis remains to be determined, however.
Divers indices cliniques, cognitifs, neurophysiologiques attestent d’atteintes céré- brales concomitantes des symptômes schizophréniques : signes neurologiques mineurs, troubles attentionnels, anomalies de la poursuite oculaire également retrouvés chez des parents indemnes de schizophrénie. Les déterminants du passage de la condition de vulnérabilité au trouble avéré demeurent mal identifiés alors qu’il serait tellement utile de les connaître.
Les schizophrénies, maladies du cerveau
L’idée d’une atteinte cérébrale chez les patients schizophrènes était avancée par Kraepelin ou Bleuler ; elle est aujourd’hui confirmée par les travaux neuropathologiques et les études en imagerie alors que sur le plan clinique des anomalies neurologiques, certes « mineures » et peu spécifiques, sont de mieux en mieux décrites : symptômes pyramidaux ou extra pyramidaux, troubles de la coordination, de l’équilibre, de la latéralisation, de l’intégration sensorielle, apraxies [8, 9]. De même des troubles cognitifs hétérogènes sont observés : attentionnels, mnésiques, fluence verbale, fonctions exécutives et troubles du langage…[1, 2, 3, 10, 11]. Enfin des particularités « neurophysiologiques » ont été décrites telles que anomalies de la poursuite oculaire, témoignant d’un déficit du monitoring interne et d’un déficit d’inhibition [12], anomalies de l’onde P 50 [13, 14], qui correspondraient à un défaut de « filtrage » sensoriel ou encore, anomalies de l’onde N300 [15].
Les données issues de l’imagerie structurale [16] révèlent des anomalies cérébrales précoces souvent modérées : atrophie corticale (plus particulièrement cortex frontal, temporal et entorhinal) ou sous corticale (striatum, thalamus, cervelet), élargissement des ventricules, anomalies de la gyration [17]. Ces anomalies pourraient précéder l’éclosion des symptômes et progresseraient peu une fois la maladie
déclarée, à l’inverse de ce qui est observé dans les troubles neuro-dégénératifs [18, 19].
L’observation d’une réduction d’environ 10 % de l’épaisseur du cortex, visible en imagerie et en post-mortem, a été largement répliquée [4, 5, 20]. La diminution de volume au niveau du cortex préfrontal pourrait être liée soit à une réduction de la taille des neurones, probablement en rapport avec une baisse du volume dendritique et axonal, soit à une réduction du nombre ou du volume des cellules gliales. Ces modifications pourraient aussi être secondaires à une perte de fonctionnalité des connections ou à une anomalie de l’agencement des couches corticales au cours du développement, notamment du fait de la localisation ectopique de certains neurones.
Les « signes neurologiques mineurs » pourraient être associés au syndrome déficitaire de certaines schizophrénies ou à des anomalies cognitives. Notre équipe a montré que les signes neurologiques mineurs sont corrélés à la dimension de désorganisation aussi bien chez des patients traités que des patients sans traitement neuroleptique et à l’existence d’anomalies physiques mineures [22] Les mêmes observations sont fréquentes chez des sujets à haut risque de schizophrénie en particulier des enfants de patients [21, 22, 14, 18, 23]. La transmission au sein des familles de patients schizophrènes a même été montrée pour certaines anomalies neurologiques [24], structurales [25, 26] ou neuro-physiologiques [12, 13, 14]. Une étude en imagerie a montré que des apparentés sains et sans déficit cognitif activent des zones cérébrales différentes de témoins sains lors d’une tâche de fonctions exécutives [27]. Tous ces indices pourraient donc être marqueurs d’un « terrain » de vulnérabilité.
A quoi correspond la « transition » psychotique [28], passage de la vulnérabilité à la pathologie ? S’agit-il d’une atteinte de certaines zones cérébrales spécifiquement responsables d’une évolution vers la schizophrénie ou d’anomalies davantage marquées chez les futurs schizophrènes ? Des facteurs de l’environnement sont-ils capables de précipiter la transition ? Une étude d’imagerie structurale rapporte en effet une diminution du volume de la substance grise lors d’un premier épisode psychopathologique chez les sujets évoluant ensuite vers une schizophrénie, contrairement à ceux qui évolueront vers un trouble affectif [29].
Les schizophrénies, maladies neuronales ?
Des anomalies dans l’expression soit de facteurs impliqués dans la transmission synaptique, la plasticité, la reconnaissance cellulaire, soit de protéines « structurales » ont été montrées par des études en immunocytochimie, des mesures de mRNA spécifiques [voir la métanalyse réalisée sur les pièces anatomiques issues de la Stanley Foundation, [29] et plus récemment grâce aux expériences de « criblage » d’expression de plusieurs milliers de gènes [30, 31, 32]. S’agit-il de causes ou de conséquences des déficits fonctionnels ou structuraux (forme et taille des dendrites,
taille des cellules) ? Un possible biais induit par les traitements, les conditions de vie, le manque d’activité socio-professionnelle est possible. Enfin, le problème de l’hétérogénéité clinique est crucial pour l’interprétation de résultats issus d’échantillons souvent réduits, parfois « poolés » pour améliorer la puissance de détection.
Ces réserves méthodologiques posées, certaines pistes émergent des données de la littérature :
— diminution de marqueurs présynaptiques [30], de marqueurs de la myélinisation [31, 32] ou de la protéine reeline qui joue un rôle essentiel durant la corticogénèse pour la bonne mise en place des couches corticales [33] ;
— selon la plupart des études, les systèmes « effecteurs » GABA et glutamate sont altérés : nette diminution de la GAD67 et des récepteurs glutamatergiques [29].
Ces deux systèmes de neurotransmission ont un rôle dans la maturation corticale : cause ou conséquence d’un déficit en amont, ces anomalies pourraient participer à l’évolutivité du trouble au moment de la maturation pubertaire ;
— augmentation de l’expression du BDNF [brain derived neurotrophic factor] [34], ou encore de l’expression à l’âge adulte de Oct 6, facteur de transcription exprimé durant le développement embryonnaire [35].
Concernant le gène du BDNF, notre équipe a rapporté un excès de l’allèle long 172-176 chez des schizophrènes à début tardif versus schizophrènes à début précoce répondeurs aux neuroleptiques, ainsi qu’un excès de l’allèle Bal 1 sur le gène du D -R [36]. Ces deux anomalies génétiques pourraient avoir un résultat commun :
3 l’hyperexpression du récepteur D . A côté de la diversité clinique intervient donc 3 une diversité génétique qui peut correspondre à des facteurs de protection ou au contraire de vulnérabilité, de réponse ou au contraire de résistance aux thérapeutiques.
Vers une nouvelle stratégie des études génétiques
Les principaux écueils pour l’identification de facteurs génétiques impliqués dans la transmission d’un trouble schizophrénique sont l’hétérogénéité des syndromes cliniques, l’implication de plusieurs gènes de pénétrance variable, interagissant entre eux et avec des facteurs environnementaux. Le fait que ces gènes soient vraisemblablement fréquents dans la population générale [37] est un élément de difficulté supplémentaire. Les travaux génétiques des dernières années ont consacré l’échec des méthodes classiques d’étude génétique.
Les stratégies actuelles de recherche d’une concordance phénotype-génotype portent sur des marqueurs phénotypiques autres que la maladie elle-même :
— défaut de modulation de l’onde P50 lors de potentiels évoqués auditifs en liaison avec un gène codant pour une sous unité du récepteur nicotinique [12, 6] ;
— liaison d’anomalies de la poursuite et des antisaccades avec un polymorphisme du gène du récepteur D3 [39] ;
— association entre un polymorphisme du gène du BDNF et la qualité de la mémoire épisodique [40].
— polymorphisme du gène de la Catéchol-O-Méthyl Transférase (COMT) (enzyme de dégradation des catecholamines) lié aux performances au test de Wisconsin de patients schizophrènes et leurs apparentés [38].
Mais il reste possible par exemple que ces polymorphismes de la COMT influencent le fonctionnement frontal [41] sans être en cause dans le processus schizophrénique, sinon d’aggraver des dysfonctions cognitives déjà altérées par un processus physiopathologique… qu’il reste à découvrir.
De nouvelles recherches se développent vers des gènes pour des protéines « inattendues », par exemple, la dysbindin [DNTB] qui aurait une activité biologique en rapport avec les protéines du cytosquelette [42], la protéine RGS4 (régulatrice des G protéines) ou encore la neuréguline [NRγ [43].
Les symptômes prémorbides et l’entrée dans la maladie
La moitié des futurs patients schizophrènes ont présenté des troubles du comportement et de l’adaptation précédant de près de dix ans la première hospitalisation :
anxiété, humeur dépressive, perte d’énergie, difficultés scolaires, agressivité, retrait, conduites suicidaires (14 fois plus que la population générale) et toxicomaniaques (environ 40 % des patients). La reconnaissance du caractère pathologique de cette période est souvent difficile, surtout quand le changement est progressif et d’autant que ces symptômes surviennent durant l’adolescence.
Divers entretiens semi-structurés peuvent être utilisés pour l’identification des symptômes prodromiques : en particulier le SIPS (interview structuré pour les symptômes prodromaux) conçu par l’équipe de McGlashan aux USA [47] et le CAARMS (Comprehensive Assessment of At Risk Mental State) développé par l’équipe de McGorry en Australie [48] A partir de ces deux outils très proches, ont été définies des situations de « très haut risque » de transition vers un état psychotique caractérisé : 30 à 40 % des patients répondant à ces critères ont développé un trouble schizophrénique dans l’année [49, 50]. Une limite au concept de prodrome est l’existence de ‘‘ faux positifs ’’ (Eaton et al. , 1995), ces tableaux pauci symptomatiques n’évoluant pas tous vers un trouble schizophrénique. Le terme « d’état mental à risque » paraît donc plus adapté [51].
Evolution diachronique des troubles : Certaines personnes présentant des signes de « vulnérabilité », (caractéristiques non « pathologiques ») vont présenter des signes « prodromiques », précédant la phase de transition vers le premier épisode psychotique (profil a). Aucune de ces transitions n’est inéluctable. En particulier, la plupart des sujets « vulnérables » n’évolueront jamais vers un état pathologique. De même, certains sujets présentant des signes « prodromiques » n’évolueront pas vers un état psychotique constitué (profil b).
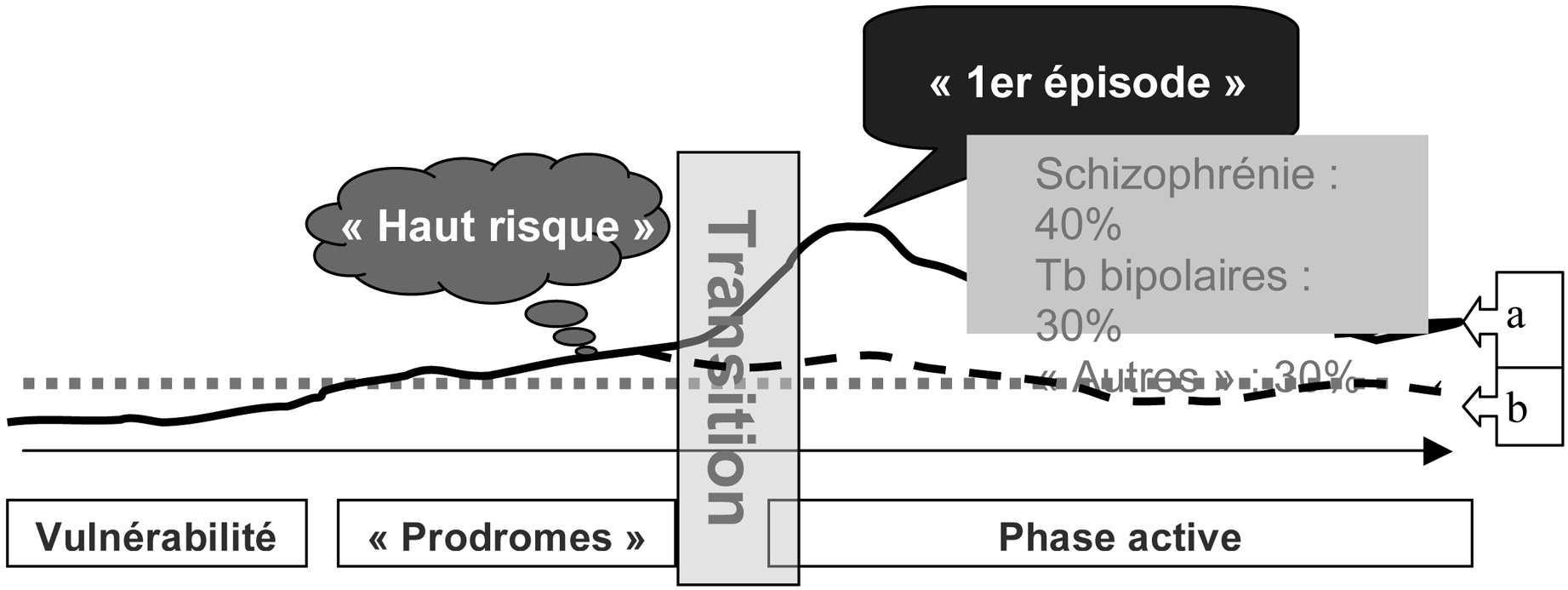
La durée de la période de psychose non traitée (Duration of Untreated Psychosis, DUP) serait un facteur déterminant la qualité de la réponse thérapeutique à long terme [52, 53]. Or, les modalités actuelles d’accès aux soins ne permettent une prise en charge adéquate en moyenne qu’un à deux ans après le début des troubles. Sur notre propre file active, nous retrouvons une durée entre le début du premier épisode psychotique et le premier traitement médicamenteux de 1,9 fi 4,2 ans et un délai de 4.2 fi 5.9 depuis le premier contact (médecin généraliste, psychologue, psychiatre) pour raison psychologique.
Certains auteurs soutiennent la pertinence de mettre en œuvre des traitements avant même que la psychose ne soit installée chez des sujets à « très haut risque de psychose » [54]. Cependant, les critères définissant les patients devant être traités et la nature même des traitements à proposer (neuroleptiques, antidépresseurs, thérapie cognitivo-comportementale, mesures éducatives…) restent à définir.
Conclusions.
La vulnérabilité aux troubles schizophréniques trouve son origine dans des anomalies du développement, l’évolution de la vulnérabilité vers la maladie n’étant pas inéluctable. Peut-on détecter précocément les sujets évoluant vers cette transition ?
Peut-on prévenir cette évolution ? On suspecte le rôle précipitant du « stress » et de certaines substances comme le cannabis [7]. En cela, le trouble schizophrénique pourrait être une maladie de l’adaptation. La mise en évidence récente des processus sous tendant la plasticité du cerveau offrent des substrats biologiques potentiels pour comprendre le fonctionnement de ce « cerveau adaptatif ».
BIBLIOGRAPHIE [1] BURGLEN F., MARCZEWSKI P., MITCHELL K.J., VAN DER LINDEN M., JOHNSON M., DANION J.M.
and al . — Impaired performance in a working memory binding task il patients with schizophrenia.
Psychiatry Res ., 2004, Mar 15, 125 (3), 247-55.
[2] DANION J.M., KAZES M., HURON C., KARCHOUNI N. — Do patients with schizophrenia consciously recollect emotional events better than neutral events ? Am. J. Psychiatry , 2003, Oct, 160 (10), 1879-81.
[3] ZALLA T., JOYCE C., SZOKE A., SCHURHOFF F., PILLON B., KOMANO O. and al . — Executive dysfunctions as potential markers of familial vulnerability to bipolar disorder and schizophrenia. Psychiatry Res ., 2004, Jan 1, 121 (3), 207-17.
[4] PAILLERE-MARTINOT M., CACLIN A., ARTIGES E., POLINE J.B., JOLIOT M., MALLET L. and al . —
Cerebral gray and white matter reductions and clinical correlates in patients with early onset schizophrenia Schizophr. Res ., 2001, May 30, 50 (1-2), 19-26.
[5] LE PROVOST J.B., BARTRES-FAZ D., PAILLERE-MARTINOT M.L., ARTIGES E., PAPPATA S., RECASENS C. and al . — Paracingulate sulcus morphology in men with early-onset schizophrenia.
Br.J. Psychiatry , 2003, Mars, 182 , 228-32.
[6] HOUY E., RAUX G., THIBAUT F., BELMONT A., DEMILY C., ALLIO G. and al. — The promoter —194 C polymorphism of the nicotinic alpha 7 receptor gene has a protective effect against the P50 sensory gating deficit. Mol Psychiatry , 2004, Mar, 9 (3), 320-2.
[7] VERDOUX H., TOURNIER M. — Cannabis use and risk of psychosis : an etiological link ?
Epidemiol Psychiatr .,Soc. 2004, Apr-Jun, 13 (2), 113-9 [8] ISMAIL B.T. et al. — Neurological abnormalities in schizophrenia : clinical, etiological and demographic correlates.
Schizophr Res., 1998, 30 , 229-238.
[9] KREBS M.O., GUT-FAYAND A., BOURDEL M.C., DISCHAMP J., OLIÉ J.P. — Validation an factoriel structure of a standardized neurological examination assessing neurological soft-signs in schizophrenia. Schizophrenia Res ., 2000, 45 , 245-60.
[10] EGAN, M.F. and al . — Relative risk for cognitive impairments in siblings of patients with schizophrenia.
Biol Psychiatry., 2001, 50 , 98-107.
[11] COMBLATT B., and al . — Cognitive and behavioral precursors of schizophrenia. Dev Psychopathol 1999, 11 , 487-508.
[12] CALKINS M.E., LACOMO W.G. — Eye movement dysfunction in schizophrenia : a heritable characteristic for enhancing phenotype definition. Am. J. Med. Genet ., 2000, Spring, 97 (l), 72-6.
[13] FREEDMAN R., COON H., MYLES-WORSLEY M., ORR-URTREGER A., OLINCY A., DAVIS A. and al . — Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc.
Natl Acad. Sci ., U S A, 1997, 94 (2), 587-92.
[14] ADLER L.E., FREEDMAN R., ROSS R.G., OLINCY A., WALDO M.C. — Elementary phenotypes in the neurobiological and genetic study of schizophrenia. Biol. Psychiatry ., 1999, Jul 1, 46 (l), 8-18.
[15] GUILLEM F., BICU M., PAMPOULOVA T., HOOPER R., BLOOM D., WOLF M.A. et al. — The cognitive and anatomo-functional basis of reality distortion in schizophrenia : a view from memory event-related potentials. Psychiatry Res ., 2003, Feb 15, 117 (2), 137-58.
[16] WRIGHT I.C., RABE-HESKETH S., WOODRUFF P.W., DAVID A.S., MURRAY R.M., BULLMORE E.T.
— Meta-analysis of regional brain volumes in schizophrenia. Am. J. Psychiatry, 2000, 157 (l), 16-25.
[17] VOGELEY K., TEPEST R., PFEIFFER U., SCHNEIDER-AXMANN T., MAIER W., HONER W.G. and al . —
Right frontal hypergyria differentiation in affected and unaffected siblings from families multiply affected with schizophrenia : a morphometric mri study. Am. J. Psychiatry , 2001, Mar, 153 (3), 494-6.
[18] SZESZKO P.R., GUNNING-DIXON F., ASHTARI M., SNYDER P.J., LIEBERMAN J.A., BILDER R.M. — Reversed cerebellar asymmetry in men with first-episode schizophrenia. Biol. Psychiatry , 2003,
Mar l, 53 (5), 450-9.
[19] KUBICKI M., SHENTON M.E., SALISBURY D.F., HIRAYASU Y., KASAI K., KIKINIS R. and al . —
Voxelbased morphometric analysis of gray matter in first episode
Schizophrenia. Neuroimage , 2002, Dec, 17 (4), 1711-9.
[20] HARRISON P.J. — The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain , 1999, Apr, 122 , 593-624.
[21] DAZZAN P., MURRAY R.M. — Neurological soft signs in first-episode psychosis : a systematic review. Br. J. Psychiatry, Suppl. 2002, Sep, 43 , s5O-7.
[22] COMBLATT B., and al . — Cognitive and behavioral precursors of schizophrenia. Dev. Psychopa- thol., 1999, 11 ,487-508.
[23] LAWRIE, S.M. and al . — Neurodevelopmental indices and the development of psychotic symptoms in subjects at high risk of schizophrenia.
Br. J. Psychiatry, 2001, 178 , 524-530.
[24] GOURION D., GOLDBERGER C., BAYLE F.J., MILLET B., OLIÉ J.P., KREBS M.O. — Neurological soft-signs and minor physical anomalies in schizophrenia : differential transmission in families.
Schizophrenia Research, 2003, 63 , 181-187.
[25] EGAN M.F., HYDE T.M., BONOMO J.B., MATTAY V.S., BIGELOW L.B., GOLDBERG T.E. et al . —
Relative risk of neurological signs in siblings of patients with schizophrenia.
Am. J. Psychiatry , 2001 Nov, 158 (11), 1827-34.
[26] SHARMA T., LANCASTER E., SIGMUNDSSON T., LEWIS S., TAKEI N., GURLING H. and al . — Lack of normal pattern of cerebral asymmetry in familial schizophrenic patients and their relatives-The Maudsley Family Study. Schizophr. Res ., 1999, Nov 30 ,40 (2), 111-20.
[27] CALLICOTT J.H., EGAN M.F., MATTAY V.S., BERTOLINO A., BONE A.D., VERCHINKSI B. and al. —
Abnormal fMRI response of the dorsolateral prefrontal cortex in cognitively intact siblings of patients with schizophrenia. Am. J. Psychiatry , 2003, Apr, 160 (4), 709-19.
[28] WEINBERGER D.R., MCCLURE R.K. — Neurotoxicity, neuroplasticity, and magnetic resonance imaging morphometry : what is happening in the schizophrenic brain ? Arch. Gen. Psychiatry , 2002, Jun, 59 (6), 553-8.
[29] KASAI K., SHENTON M.E., SALISBURY D.F., HIRAYASU Y., LEE C.U., CISZEWSKI A.A. and al . —
Progressive decrease of left superior temporal gyrus gray matter volume in patients with first-episode schizophrenia. Am. J. Psychiatry , 2003 Jan, 160 (1), 156-64.
[30] KNABLE M.B., BARCI B.M., BARTKO J.J., WEBSTER M.J., TORREY E.F. — Molecular abnormalities in the major psychiatric illnesses : Classification and Regression Tree [CRT] analysis of post-mortem prefrontal markers. Mol. Psychiatry , 2002, 7 , 392-404.
[31] MIRNICS K., MIDDLETON F.A., LEWIS D.A., LEVITT P. — Analysis of complex brain disorders with gene expression microarrays : schizophrenia as a disease of the synapse. Trends Neurosci ., 2001, 24 , 479-86.
[32] HAKAK Y., WALKER J.R., LI C., WONG W.H., DAVIS K.L., BUXBAUM J.D., HAROUTUNIAN V. and al . — Genomewide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia.
Proc. Natl Acad. Sci., U S A, . 2001, 98 , 4746-51.
[33] VAWTER M.P., CROOK J.M., HYDE T.M., KLEIIUNAN J.E., WEINBERGER D.R., BECKER K.G. and al . — Microarray analysis of gene expression in the prefrontal cortex in schizophrenia : a preliminary study.
Schizophr. Res ., 2002, 58 , 11-20.
[34] BAR I., LAMBERT DE ROUVROIT C., GOFFINET A.M. — The evolution of cortical development.
An hypothesis based on the role of the Reelin signaling pathway. Trends Neurosci., 2000, 23 , 633-8.
[35] TAKAHASHI M., SHIRAKAWA 0., TOYOOKA K., KITAMURA N., HASHIMOTO T., MAEDA K. and al . — Abnorinal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients.
Mol. Psychiatry , 2000, May, 5 (3), 293-300.
[36] ILIA M., BEASLEY C., MEIJER D., KERWIN R., COTTER D., EVERALL I. and al. — Expression of
Oct-6, a POU III domain transcription factor, in schizophrenia.
Am. J. Psychiatry , 2002, Jul, 1 59 (7), 1174-82.
[37] KREBS M.O., GUILLIN O., BOURDELL M.C., SCHWARTZ J.C., OLIÉ J.P., POIRIER M.F. and al . —
Brain derived neurotrophic factor [BDNF] gene variants association with age at onset and therapeutic response in schizophrenia. Mol. Psychiatry , 2000, 5 , (5), 558-62.
[38] GERSHON E.S. — Bipolar illness and schizophrenia as oligogenic diseases : implications for the future. Biol. Psychiatry , 2000, 47 (3), 240-4.
[39] EGAN M.F., GOLDBERG T.E., KOLACHANA B.S., CALLICOTT J.H., MAZZANTI C.M., STRAUB R.E., and al . — Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia.
Proc. Natl Acad. Sc., USA, 2001, 98 , (12), 6917-22.
[40] RYBAKOWSKI J.K., BORKOWSKA A., CZERSKI P.M., HAUSER J. — Dopamine D3 receptor [DRD3] gene polymorphism is associated with the intensity of eye movement disturbances in schizophrenic patients and healthy subjects. Mol. Psychiatry, 2001, 6 (6), 718-24.
[41] EGAN M.F., KOJIMA M., CALLICOTT J.H., GOLDBERG T.E., KOLACHANA B.S., BERTOLINO A. and al . — The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function.
Cell , 2003, Jan 24, 112 (2), 257-69.
[42] WEINBERGER D.R., EGAN M.F., BERTOLINO A., CALLICOTT J.H., MATTAY V.S., LIPSKA B.K. and al. — Prefrontal neurons and the genetics of schizophrenia. Biol. Psychiatry , 2001 Dec 1, 50 (11), 825-44.
[43] STRAUB R.E., JIANG Y., MACLEAN C.J., MA Y., WEBB B.T., MYAKISHEV M.V. and al. — Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet , 2002, Aug.,71 (2), 337-48. Epub 2002 Jul 03.
Erratum in :
Am. J. Hum. Genet., 2002, Oct, 72 (4), 1007.
[44] HARRISON P.J., OWEN M.J. — Genes for schizophrenia ? Recent findings and their pathophysiological implications. Lancet , 2003, 361 , 417-9.
[45] BAXTER D., APPLEBY L. — Case register study of suicide risk in mental disorders.
Br. J.
Psychiatry, 1999, 175, 322-6 [46] DERVAUX A., BAYLE F.J., KREBS M.O. — Substance misuse in schizophrenic patients : similarities and differences between UK and France. Br. J.Psychiatry, 2002, 180 ,381 [47] MILLER T.J., MCGLASHAN T.H., ROSEN J.L., SOMJEE L., MARKOVICH P.J., STEIN K. and al . —
Prospective diagnosis of the initial prodrome for schizophrenia based on the Structured Interview for Prodromal Syndromes : preliminary evidence of interrater reliability and predictive validity. Am. J. Psychiatry, 2002, 159 (5), 863-5.
[48] MCGORRY P.D., YUNG A.R., PHILLIPS L.J., YUEN H.P. — The « close-in » or ultra high-risk model : a safe and effective strategy for research and clinical intervention in prepsychotic mental disorder. Schizophr. Bull., 2003, 29 , 771-90.
[49] KILLER, T.J., MC GLASHAN T.H., WOODS S.W. and al . — Symptom assessment in schizophrenic prodromal states.
Psychiatra , 1999, 70 , 273-287.
[50] YUNG A.R., PHILLIPS L.J., YUEN H.P., MCGORRY P.D. — Risk factors for psychosis in an ultra high-risk group : psychopathology and clinical features. Schizophr. Res ., 2004, Apr 1, 67 (2-3), 131-42.
[51] YUNG A.R., MC GORRY P.D. — The prodromal phase of first episode psychosis : past and current conceptualisations. Schizophrenia Bull., 1996, 22 , 353-70.
[52] LIEBERMAN J.A., PERKINS D., BELGER A. and al . — The early stages of schizophrenia : speculations on pathogenesis, pathophysiology, and therapeutic approaches.
Biol. Psychiatry , 2001, 50 (11), 884-97.
[53] CANCEIL O., SAMPAIO-MEIRELES M., BOURDEL M.C., OLIÉ J.P., POIRIER M.F. — Psychopathology, Social Adaptation And Quality Of Life In Schizophrenia : Study Methodology With Baseline Analysis Of A 145 Patients Cohort Study. Soumis pour publication à Psychiatry Research .
[54] HARRIGAN S.M., MCGORRY P.D., KRSTEV H. — Does treatment delay in first-episode psychosis really matter ? Psychol. Med., 2003, 33 (1), 97-110.
DISCUSSION
M. Gilbert LELORD
Les résultats suivants ont-ils été confirmés par plusieurs laboratoires : défaut de modulation des potentiels auditifs (onde P50) et particularités du gène nicotinique (alpha 7), anomalies de la poursuite oculaire et particularités du gène du récepteur D3, ou bien ces résultats sont-ils encore isolés ?
Les résultats que j’ai mentionnés sont des résultats qui ont été reproduits, en particulier ceux qui indiquent un défaut de modulation et des anomalies des saccades oculaires en relation avec les particularités du gène du récepteur D3.
M. Pierre PICHOT
Dans votre exposé, vous avez rappelé une série de faits qui sont actuellement généralement admis : la schizophrénie peut être conçue suivant un modèle postulant un continuum vulnérabilité-stress, dans lequel de multiples facteurs génétiques et environnementaux agissent de manière additionnelle. On peut schématiquement distinguer dans le cadre de ce continuum les sujets apparemment sains, mais à prédisposition génétique (en particulier dans les familles de schizophrènes), éventuellement repérables par l’existence d’anomalies neuro-physiologiques, comme les troubles de la poursuite oculaire, les sujets vulnérables présentant des anomalies diverses précoces, comme l’ont montré de nombreuses études de cohortes d’enfants de parents schizophrènes, les sujets ‘‘ à très haut risque ’’ détectables avec une haute probabilité par leurs caractéristiques comportementales considérées comme prodromiques, et enfin les sujets dont les troubles mentaux sont nets. L’éventualité du passage d’un stade à l’autre est en rapport avec l’action de facteurs génétiques et de milieu, et l’existence de certains de ces derniers a suggéré des possibilités de mesures préventives. Le problème que je souhaiterais soulever est en rapport avec un détail de votre exposé. Les symptômes prodromiques définissant un très haut risque de développement d’un trouble schizophrénique ne sont pas absolument spécifiques, ils indiquent aussi un risque pour le développement d’un trouble bipolaire. La nosologie actuelle des psychoses admet, dans ses grandes lignes, la dichotomie créée il y a un peu plus d’un siècle par Kraepelin, opposant la démence précoce —notre actuelle schizophrénie- et la psychose maniaco-dépressive —pour l’essentiel, notre actuel trouble bipolaire. Cette conception, considérée longtemps comme un dogme, est aujourd’hui contestée. L’existence de limites rigides entre les deux entités est contestée, sur le plan clinique (par l’existence des troubles schizo-affectifs), comme sur le plan biologique (par les résultats des études anatomiques de neuro-physiologie fonctionnelle) et sur le plan génétique. Tout se passe comme s’il existait un processus pathologique commun, que l’on peut si l’on veut, qualifier de psychotique, et des processus spécifiques,
orientant vers l’une ou l’autre psychose. Ainsi, dans un très récent article au titre provocateur ‘‘ Le début de la fin de la dichotomie kraepelinienne ’’, Craddock a suggéré qu’il existe des gènes spécifiques pour la schizophrénie, des gènes spécifiques pour le trouble bipolaire, mais aussi des gènes correspondant à la fois à un risque pour la schizophrénie, le trouble schizo-affectif et le trouble bipolaire. Leur combinaison entraîne l’existence d’un spectre de tableaux cliniques, que l’on peut schématiquement représenter d’une manière linéaire. Aux deux extrémités de cette distribution se situent ce que l’auteur appelle la schizophrénie et le trouble bipolaire ‘‘ prototypaux ’’ mais la majorité des cas se situe dans une position intermédiaire. Cette conception concernant les facteurs génétiques rappelle celle proposée, il y a déjà plusieurs décennies, au Royaume-Uni par Kendell qui, condensant par des méthodes statistiques la symptomatologie schizophrénique et affective en un modèle dimensionnel, affirmait qu’en fait l’ensemble des sujets psychotiques pouvaient être représentés en fonction de leurs symptômes propres sur cette dimension et que la répartition des malades psychotiques avait une allure plus ou moins gaussienne, c’est-à-dire que le plus grand nombre de sujets avait une symptomatologie ‘‘ mixte ’’. Puis-je demander aux auteurs leur position vis-à-vis de ces idées qui rejoignent, sous une forme actuelle, celle de la ‘‘ psychose unique ’’, défendue entre autres par Guislain et Griesinger au XIXe siècle, et dans quelle mesure ces idées sont compatibles avec les données des travaux récents et leurs propres recherches ?
Votre connaissance des aspects nosologiques ne m’autorise pas à vous proposer une réponse à la question fondamentale que vous posez : continuum au sein d’un ensemble psychotique unique ou hétérogénétique ou des psychoses ? Les résultats les plus actuels montrent que certaines des anomalies, cérébrales neuronales, électrophysiologiques ou moléculaires sont communes au trouble schizophrénique et au trouble bipolaire : on retrouve ces anomalies dans ces deux contextes psychopathologiques. Comme vous le savez, se pose la même question aujourd’hui d’un continuum entre maladies dépressives et maladies neurodégénératives. Tout ceci pourrait tendre à nous ramener vers la notion de psychose unique au sein de laquelle existeraient des caractéristiques endophénotypiques pouvant nous conduire dans le futur vers de nouvelles modalités de classification.
M. Pierre LEFÈBVRE
Le dysfonctionnement neuronal dont parle Jean-Pierre Olié me paraît tout à fait important.
Il peut expliquer les coenesthésies à l’origine des syndromes de dépersonnalisation observés dès les débuts de la schizophrénie. Ils expliquent certains vécus de transformation (qui se traduisent en particulier par le signe du miroir) et qui s’accompagnent de symptômes neurologiques (parasitisme mimique, rires immotivés, dystonies…).
J’adhère tout à fait à votre hypothèse selon laquelle certains dysfonctionnements neuronaux pourraient participer à l’éclosion de syndromes de dépersonnalisation inauguraux de la schizophrénie M. Pierre RONDOT
Les examens présentés permettent-ils de différencier les épisodes psychotiques aigus de l’adulte jeune et le début d’une schizophrénie ?
Les examens présentés ne sont actuellement pas entrés dans la pratique clinique. L’espoir est bien d’aller vers l’identification de paramètres prédicteurs d’une possible transition psychotique.
M. François-Bernard MICHEL
L’approche scientifique du sujet, par Jean-Pierre Olié, est particulièrement précieuse car le profane que je suis est inquiet de constater la fréquence de diagnostics de schizophrénies portés sur des arguments cliniques et des enfants jeunes (12-14 ans). N’y a-t-il pas un risque de diagnostic par excès ?
Votre question est particulièrement difficile dans la mesure où le diagnostic de schizophrénie ne peut être porté qu’à partir du moment où l’évolution est incontestablement devenue chronique. Et pourtant tout l’enjeu est d’anticiper cette éventuelle chronicité en mettant au point des thérapeutiques préventives. Cependant nous n’en sommes pas encore là, même si les nouvelles thérapeutiques, médicamenteuses ou psychologiques, sont aujourd’hui mieux utilisables (en particulier grâce à la meilleure tolérance de nouveaux médicaments antipsychotiques) et donc plus facilement mises en œuvre dès le constat d’une menace d’évolution psychotique chronique.
M. Michel HAMON
Je crois savoir que la prévalence de la schizophrénie est remarquablement stable d’un pays à l’autre, de l’ordre de 1 %. Quelles interprétations peut-on en donner, notamment au regard de l’interaction gène-environnement dans l’étiopathogénie de la maladie ? De nouveaux antipsychotiques sont apparus ces dernières années, aux mécanismes d’action de mieux en mieux identifiés. L’analyse de leurs effets peut-elle contribuer à affiner les connaissances sur la physiopathologie de la schizophrénie ? Enfin, la neuro-imagerie, avec la caméra à positons et l’IRM fonctionnelle, semble beaucoup contribuer à identifier les anomalies du fonctionnement cérébral dans diverses pathologies du système nerveux central. Où en est-on dans le cas de la schizophrénie, et que peut-on encore en attendre ?
Le taux de prévalence des schizophrénies est effectivement stable à travers le monde :
les modalités d’expression sont différentes. Pour l’heure, il ne me semble pas que l’on puisse affirmer que les nouveaux médicaments antipsychotiques, qui sont à mes yeux un progrès incontestable, permettent d’affiner les connaissances sur la physiopathologie des schizophrénies. Par contre, les techniques d’imagerie n’ont pas fini de nous faire progresser dans l’indentification des anomalies précédant ou accompagnant les schizophrénies.
M. Jean-François ALLILAIRE
La pratique clinique montre la très grande fréquence de la consommation et l’abus de cannabis chez les patients schizophrènes, à la fois comme comorbidité à la phase d’état de la maladie, mais aussi pendant la phase pré-clinique du trouble et enfin et peut-être surtout dans les mois et parfois semaines qui précèdent l’éclosion du trouble, au point qu’on a pu parler de véritable ‘‘ pharmacopsychose ’’ suivant l’expression qui avait été proposée par Pierre Deniker. De plus, on sait par les enquêtes épidémiologiques que la consommation de cannabis doit être considérée comme un facteur de risque pour l’apparition du trouble schizophrénique. Au-delà du lien statistique mis en évidence par les études épidémiologiques, y a-t-il un lien de causalité scientifiquement et formellement démontré à ce jour par les recherches actuelles ? Et si c’est le cas, quel en serait le mécanisme neurobiologique ?
Nous connaissons la forte prévalence de consommation et d’abus de cannabis chez les schizophrènes (environ 40 % d’entre eux ont consommé ou consomment du cannabis) ;
on sait également qu’il existe une vulnérabilité particulière aux effets psychodysleptiques du cannabis chez certains d’entre eux ce qui pourrait conduire à un élargissement du concept de pharmacopsychose ; certains arguments sont enfin en faveur de l’existence d’une période de vulnérabilité spécifique aux effets psychotisants du cannabis à la fin de la maturation neuronale c’est à dire vers l’adolescence et le début de l’âge adulte. Le mécanisme par lequel le cannabis pourrait révéler ou faciliter des symptômes psychotiques est certainement une hyperdopaminergie mésolimbique.
M. André VACHERON
Le défaut de filtrage sensoriel serait rétabli par la nicotine. Le tabagisme aurait-il une action favorable chez les schizophrènes ?
On sait effectivement que les schizophrènes surtout hommes ont une consommation de tabac nettement supérieure à celle de la population générale. S’agit-il d’une auto-thérapie du filtrage sensoriel ? En tout cas, il n’est pas démontré que ce soit une thérapeutique des symptômes schizophréniques. Ceci nous montre peut être que le traitement d’un indice endophénotypique cognitif ou électrophysiologique n’est pas nécessairement suffisant pour traiter l’état pathologique constitué.
* SHU/ E0117 INSERM- Hôpital Ste Anne- Université PARIS V, 1 rue Cabanis — 75674 Paris cedex 14. ** Membre de l’Académie nationale de médecine Tirés à part : Professeur Jean-Pierre OLIÉ, même adresse. Article reçu le 30 juillet 2004, accepté le 28 février 2005.
Bull. Acad. Natle Méd., 2005, 189, no 5, 935-947, séance du 17 mai 2005