
Résumé
L’initiation de la puberté est un processus complexe caractérisé par l’augmentation de la sécrétion de la GnRH entraînant le réveil de l’axe gonadotrope. Récemment, des avancées majeures dans la compréhension de l’initiation de la puberté ont été réalisées par l’étude des déterminants génétiques de la puberté normale et la description de mutations perte de fonction dans plusieurs gènes codants pour des neuropeptides (kisspeptins, neurokinin B) ou leurs récepteurs. Un réseau de neurones hypothalamiques contrôlant la réactivation de l’axe gonadotrope est en cours de caractérisation. La chronologie de cette activation pourrait dépendre d’une régulation complexe par la protéine Lin28 qui est codée par un gène hétérochronique. La poursuite de la caractérisation des gènes des maladies de la puberté va permettre de compléter la description de ce réseau. Les conséquences de ces travaux sont doubles : une meilleure compréhension de l’une des dernières énigmes en physiologie et la description de nouvelles cibles thérapeutiques en reproduction.
Summary
Puberty is triggered by a complex neuroendocrine mechanism that leads to an increase in GnRH release at the end of the childhood and, hence, to reactivation of the gonadotropic axis. Recent human genetic studies have led to major breakthroughs in our understanding of puberty onset. A network of hypothalamic neurons controlling GnRH release has just been characterized. It appears that the timing of puberty onset is under the control of the heterochronic gene Lin28, which encodes a protein regulating microRNA maturation. Characterization of additional gene defects associated with abnormal puberty onset is needed to further characterize this neuroendocrine network and to identify new therapeutic targets for reproductive disorders.
INTRODUCTION
La puberté, dont la durée est d’environ quatre ans, correspond à un ensemble de phénomènes maturatifs neuroendocriniens complexes qui amènent un individu à un stade de développement permettant la reproduction. Elle débute en moyenne à l’âge de 10,5 ans chez la fille et à 11,5 ans chez le garçon avec des extrêmes de 9 à 13 ans chez la fille, et de 10 à 14 ans chez le garçon [1]. Elle témoigne de la réactivation progressive de la commande hypothalamo-hypophysaire de l’axe gonadotrope à la fin de l’enfance dont le mécanisme est très mal compris [2, 3].
L’initiation de la puberté est due à une augmentation importante de la sécrétion de GnRH par l’hypothalamus après une phase de quiescence pendant l’enfance [4].
Cette période de quiescence juvénile est liée à une inhibition centrale de la sécrétion de GnRH principalement par l’acide gamma-amino butyrique [5]. La levée de l’inhibition GABAergique associée à l’augmentation de l’activité des neurones glutamaergiques contribue à l’augmentation de la sécrétion pulsatile de GnRH et au démarrage pubertaire. Au fur et à mesure que la puberté progresse, d’autres neurotransmetteurs participent à la régulation de la sécrétion de GnRH (Figure 1), avec des différences notables entre les deux sexes [6]. L’influence de ces modulateurs sur la sécrétion de la GnRH est indiscutable, mais aucun d’entre eux n’a la capacité de réactiver à lui seul l’axe gonadotrope pendant la période juvénile.
Fig. 1. — L’initiation de la puberté : un réseau neuroendocrinien complexe composé d’activateurs et inhibiteurs régulant la sécrétion de la GnRH.
Les avancées les plus marquantes de ces dernières années dans la compréhension de l’initiation de la puberté ont été obtenues par la génétique humaine. Deux grandes stratégies ont été suivies par des groupes de recherche différents. La première a consisté à caractériser les déterminants génétiques de l’âge normal de la puberté par une approche d’association pan-génomique. La deuxième approche complémentaire à la première, est basée sur la caractérisation des mécanismes moléculaires des maladies génétiques de l’initiation de la puberté. La première stratégie a mis en évidence notamment le rôle de Lin28 qui participe à la maturation des microARNs alors que la seconde approche a permis d’initier la caractérisation du réseau hypothalamique participant à la réactivation de l’axe gonadotrope. La suite de cet article décrit les résultats obtenus par ces deux stratégies.
LES
ANALYSES
D’ASSOCIATIONS
PAN-GÉNOMIQUES
DE
LA
PUBERTÉ
NORMALE
L’âge de la puberté est déterminé par des facteurs génétiques. En effet, il existe une forte corrélation de l’âge de la ménarche entre les mères et les filles et 40 à 95 % de la variabilité de l’âge de la puberté dépend de facteurs génétiques héritables [7].
Plusieurs analyses pan-génomiques récentes ont montré une association entre deux loci (6q21, 9q31.2) et l’âge de la ménarche par des analyses de régression [8-11]. Le gène le plus proche des marqueurs localisés en 9q31.2 est le TMEM38B qui est une protéine membranaire dont la fonction est inconnue. Le gène le plus proche du deuxième locus 6q21 code pour la protéine Lin28b. Une association entre polymorphismes de Lin28b et la taille adulte est également décrite et ceci indépendamment de la l’âge de la ménarche [8]. Cette observation est intéressante puisque la vitesse de croissance augmente fortement pendant la puberté. Des méta-analyses sur 100 000 femmes ont confirmé ces résultats et permis de décrire de nouveaux loci dont dix sont fortement significatifs [30]. Plusieurs de ces gènes sont associés à l’indice de masse corporelle, sont situés à proximité de gènes impliqués dans l’homéostasie énergétique ou participent à la régulation de fonctions endocrines.
Ces résultats sont finalement assez logiques puisque les liens entre indice de masse corporelle, homéostasie énergétique et puberté sont connus depuis de nombreuses années. Il est certain que la mise en évidence de l’association avec Lin28b ouvre des horizons très intéressants. La fonction de cette protéine est mal connue. Elle participe à la vitesse du développement larvaire de C élégans probablement par une régulation de la maturation des microARNs [12], petits ARN non codants participant à la régulation post-transcriptionnelle de l’expression des gènes souvent associée à des processus de différentiation et plasticité neuronale. Une souris transgénique sur-exprimant Lin28a présente un retard pubertaire probablement d’origine centrale. Lin 28b semble donc participer à la maturation post-natale de l’hypothalamus probablement par une régulation de la maturation des microARNs dans l’hypothalamus.
L’IMPORTANCE DES FACTEURS DE L’ENVIRONNEMENT DANS L’INITIATION DE LA PUBERTÉ NORMALE
La part des facteurs environnementaux dans l’initiation de la puberté a aussi été suggérée par plusieurs observations dont la plus importante est la baisse de l’âge de la ménarche depuis le milieu du xixe siècle [1]. Cette évolution est essentiellement expliquée par l’amélioration des conditions de vie. L’action de perturbateurs endocriniens dont la concentration a fortement augmenté depuis le début de l’aire industrielle a également été proposée mais jamais démontrée définitivement. Les produits synthétiques les plus souvent cités sont les pesticides, des dérivés de la dioxine ou les phtalates. Certains composés naturels tels que les phytoestrogènes ont également été cités par plusieurs études. Le mécanisme d’action proposé est la fonction estrogénique ou anti-androgénique de ces composés.
Plusieurs études ont démontré une fréquence élevée de puberté avancée voire précoce chez les enfants adoptés [13, 14]. Le risque de puberté précoce est quinze à vingt fois plus élevé après adoption d’un enfant au Danemark ou quatre-vingts fois plus élevée en Belgique. Une corrélation entre l’âge de la puberté et certains perturbateurs endocriniens a été proposée chez ces enfants.
LES MALADIES DE L’INITIATION DE LA PUBERTÉ : UN MODÈLE D’ÉTUDE POUR COMPRENDRE LA RÉACTIVATION DE L’AXE GONADOTROPE
Les maladies de l’initiation de la puberté sont réparties en deux catégories. Une maturation trop tardive de l’axe gonadotrope qui conduit à un retard pubertaire défini comme l’absence de signe de puberté chez la fille à treize ans et chez le garçon à quatorze ans. Une maturation trop précoce de l’axe gonadotrope qui conduit à une puberté précoce c’est-à-dire l’apparition de caractères sexuels avant l’âge de huit ans chez la fille, neuf ans chez le garçon. Ces deux types d’anomalies sont dépendants ou non des gonadotrophines. Les maladies de la puberté peuvent être génétiques ou bien acquises. Les plus grands progrès de ces dernières années ont été réalisées sur la génétique des maladies de l’initiation de la puberté. Dans la suite de cet article de revue, nous nous intéressons uniquement aux maladies rares de l’initiation de la puberté « gonadotrophines-dépendantes », c’est-à-dire aux anomalies de la commande neuroendocrinienne de l’axe gonadotrope.
L’hypogonadisme hypogonadotrope (HH) est une maladie endocrinienne due à un dysfonctionnement de l’unité hypothalamo-hypophysaire, en rapport avec une anomalie de la synthèse, de la sécrétion ou de la capacité de la GnRH à induire la synthèse et le relargage des gonadotrophines. Le diagnostic de HH repose sur la mise en évidence dans la majorité des cas d’un taux de testostérone plasmatique bas associé à des concentrations plasmatiques normales ou basses de gonadotrophines LH et FSH. L’évaluation clinique du volume testiculaire chez l’homme ou globalement du stade de Tanner ainsi que le dosage de l’inhibine B apportent des rensei-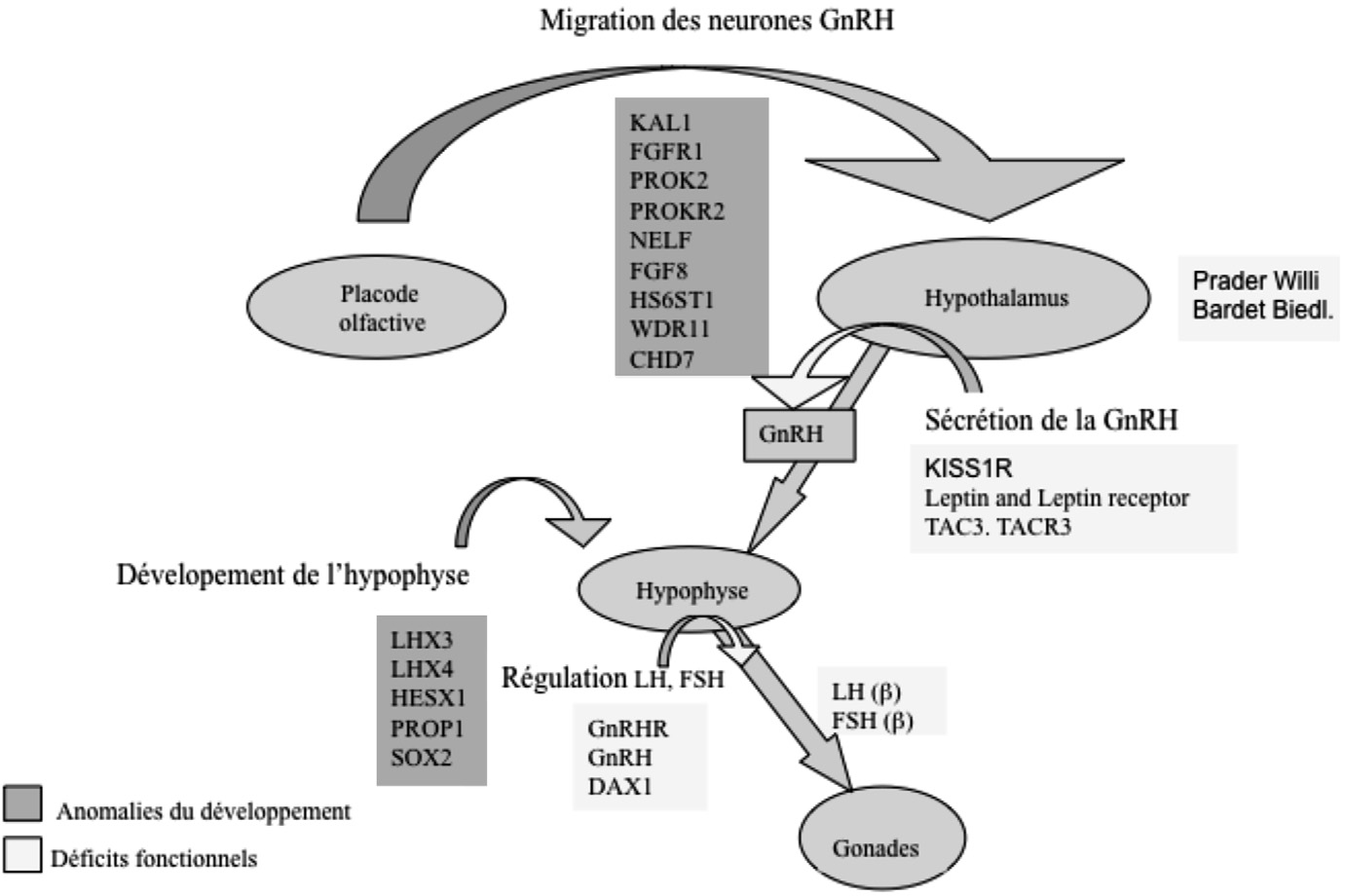 gnements complémentaires sur la profondeur de l’atteinte gonadotrope. L’HH peut être congénital ou acquis. Dans le cas des déficits gonadotropes congénitaux, plusieurs modèles de transmission sont décrits : liés au chromosome X, autosomique dominant ou récessif. L’HH peut être isolé ou associé à d’autres déficits hypophysaires et/ou malformations, dont notamment l’anosmie ce qui définie le syndrome de Kallmann (Figure 2). Lorsque le déficit gonadotrope n’est pas associé à une anosmie, une malformation ou tout autre symptôme, il est dit isolé (IHH) [15].
gnements complémentaires sur la profondeur de l’atteinte gonadotrope. L’HH peut être congénital ou acquis. Dans le cas des déficits gonadotropes congénitaux, plusieurs modèles de transmission sont décrits : liés au chromosome X, autosomique dominant ou récessif. L’HH peut être isolé ou associé à d’autres déficits hypophysaires et/ou malformations, dont notamment l’anosmie ce qui définie le syndrome de Kallmann (Figure 2). Lorsque le déficit gonadotrope n’est pas associé à une anosmie, une malformation ou tout autre symptôme, il est dit isolé (IHH) [15].
Fig. 2. — La génétique du déficit gonadotrope isolé, combiné ou syndromique. Les gènes sont indiqués en fonction du niveau d’action et du mécanisme physiopathologique, anomalie du développement (bleu) ou anomalie fonctionnelle (jaune).
L’IHH est une maladie mendélienne monogénique. Les gènes codants pour les protéines participant à l’axe gonadotrope étaient les gènes candidats les plus évidents. Depuis la première description d’une délétion du gène coant pour la GnRH, GnRH1 , au début des années quatre-vingt-dix chez la souris hypogonadique, le gène de la GnRH, est devenu rapidement un gène candidat naturel dans l’IHH [16]. Ce n’est pourtant qu’en 2009 que deux groupes ont rapporté des mutations homozygotes de GnRH1 chez l’homme [17, 18]. Le phénotype rapporté était celui d’un IHH avec déficit gonadotrope complet. Les mutations homozygotes de GnRH1 sont peu fréquentes dans l’IHH en Europe (1/150) et aux États-Unis (1/310) [17, 18]. Contrairement aux mutations de la GnRH, très peu fréquentes et récemment décrites, 22 mutations perte de fonction du récepteur de la GnRH ( GnRHR ) ont été décrites dans le domaine transmembranaire ou dans les boucles intra et extra cellulaires [19].
Les mutations de GNRHR ont été les premières identifiées dans l’IHH [20, 21] et sont à l’origine d’environ 40 % des cas d’IHH familiaux avec transmission récessive [19]. En tenant compte des cas sporadiques, la fréquence des mutations de GnRHR est évaluée entre 6 à 17 % selon les études [19]. L’IHH peut être partiel ou complet, et au sein d’une même famille, il existe différents degrés de déficit gonadotrope pour la même mutation [22, 23]. Des gènes modificateurs de l’expression du phénotype ont été évoqués ainsi qu’un modèle de digénisme [24]. La part des facteurs environnementaux dans cette variabilité de l’expression phénotypique est inconnue.
Deux découvertes ont récemment permis une avancée majeure dans la compréhension de la régulation de la sécrétion de GnRH. Elles ont été réalisées par cartographie du génome, une stratégie qui n’est pas basée sur l’hypothèse de gènes candidats et permet donc de proposer de nouveaux modèles physiopathologiques originaux.
En 2003, deux groupes indépendants ont identifié des mutations perte de fonction dans le gène KiSS1R chez des patients ayant une forme familiale d’IHH [25, 26]. Le phénotype observé est en tout point similaire à celui décrit pour les mutations de GnRHR ce qui indiquait que le déficit gonadotrope est probablement du à une anomalie de la régulation hypothalamo-hypophysaire de l’axe gonadotrope. Les modèles animaux ont depuis largement confirmé cette hypothèse. La fréquence des mutations de KISS1R est faible dans le déficit gonadotrope isolé : vingt-six cas appartenant à neuf familles différentes ont été décrits depuis 2003 [25-31]. Certains patients ont une cryptorchidie bilatérale et un micropénis à la naissance ce qui suggère que le déficit débute dés la vie fœtale [31]. La majorité des cas est transmis selon un mode autosomal récessif bien que récemment nous ayons caractérisé une transmission dominante (Chevrier et al , soumis pour publication).
De nombreux travaux ont été réalisés pour comprendre la fonction des kisspeptins hypothalamiques [32]. Il ressort de ces travaux que ces peptides sont les sécrétagogues les plus puissants de la GnRH en activant KISS1R à la surface des neurones GnRH. Ils participent au rétro-contrôle négatif et positif des estrogènes sur l’axe gonadotrope et donc explique en partie l’induction du pic ovulatoire de LH.
L’activation du système kisspeptine/KISS1R augmente à la fin de la phase juvénile par deux mécanismes complémentaires, une augmentation de la synthèse des kisspeptins associée à une réponse plus intense de KiSS1R aux kisspeptins. Les mécanismes de l’activation du système kisspeptin sont mal connus. Ils pourraient impliquer une régulation positive par les estrogènes. Nos résultats récents montrent que les kisspeptins participent à la régulation de l’axe gonadotrope durant la vie fœtale (Guimiot et al , soumis pour publication) ce qui explique le micropénis et la cryptorchidie observés chez certains enfants.
La découverte récente du rôle majeur du système kisspeptine/GPR54 dans la régulation de l’axe gonadotrope [25] a permis de montrer que ces peptides sont des puissants sécrétagogues de la GnRH dont l’activité hypothalamique augmente à la fin de la phase juvénile. Les mécanismes responsable de cette augmentation sont inconnus et sont probablement la conséquence de la maturation post-natale de l’hypothalamus et de la plasticité des neurones hypothalamiques [33]. De nombreux facteurs métaboliques périphériques dont la leptine participent également à la réactivation de l’axe gonadotrope. Ces facteurs seraient plus permissifs qu’initiateurs de la puberté mais pourraient favoriser l’augmentation de l’activité biologique des kisspeptins. Dans l’espèce humaine, les kisspeptins stimulent également la sécrétion de la LH et de la FSH chez les adultes, probablement par une action directe sur les neurones GnRH. Le lien entre kisspeptins et initiation de la puberté a été définitivement confirmé par la description récente d’une mutation de KISS1 dans une famille turque Kurde consanguine entrainant chez le propositus (une jeune fille de quinze ans) et trois de ses sœurs, une absence de puberté et un hypogonadisme hypogonadotrope sans anosmie [34].
Par une approche similaire de cartographie du génome, Topaloglu et collaborateurs ont mis en évidence des mutations homozygotes de deux gènes inattendus chez des patients ayant une forme familiale d’IHH [35] : le gène codant pour la neurokinine B ou TAC3 (MIM : 162330) et de son récepteur, TACR3 (MIM : 162332). Plusieurs articles ont depuis rapporté des mutations inactivatrices de
TAC3 et TACR3 dans de nombreux cas familiaux [36]. Le phénotype ne comprend pas d’anosmie, le déficit gonadotrope est toujours isolé et la transmission la plus fréquente est récessive mais de nombreuses mutations hétérozygotes sont décrites. Une analyse plus fine des phé- notypes montre des divergences notables entre les cas publiés qui pourraient dépendre des mutations mais également des modes d’investigation clinique et paraclinique de ces patients ainsi des antécédents de traitement avant la première investigation clinique. Le phénotype semble être fréquemment réversible à l’âge adulte.
Depuis deux ans, les études sur les modèles animaux ont permis de mieux décrire le rôle de ces deux nouveaux acteurs de l’axe gonadotrope. Les différentes études réalisées chez le rongeur, le singe ou le mouton mettent en évidence un réseau de neurones exprimant les kisspeptins et la Neurokinin B et leurs récepteurs respectifs dans le noyau arqué. Ces neurones sont interconnectés et envoient des projections dans l’éminence médiane vers les axones des neurones à GnRH. De plus, les neurones kisspeptines coexpriment la Neurokinin B et la dynorphine. L’administration centrale d’agoniste des récepteurs de la Neurokinin B induit un relargage de LH chez le mouton, le singe, le rat et la souris [37-39]. Par ailleurs, l’expression de KISS1 et TAC3 sont régulés négativement par les estrogènes : cela suggère fortement que les neurones kisspeptines comme les neurones Neurokinin B sont les relais de l’effet négatif de l’estradiol sur la secrétion de la GnRH. Cependant, les modèles de souris n’exprimant pas KISS1R sont infertiles alors que les souris n’exprimant pas TACR3 ont une fertilité normale [26, 40-42]. Ces différences suggèrent que la Neurokinin B joue un rôle activateur sur l’axe gonadotrope complémentaire mais pas identique aux kisspeptins.
Des mutations inactivatrices ont été décrites dans d’autres gènes participant directement à l’axe gonadotrope comme la sous unité β de la LH (MIM : 152780) ou de la FSH (MIM : 136530), dans de très rares cas de déficit gonadotrope. Cette hypothèse est évoquée devant une dissociation nette des concentrations sériques de la LH et de la FSH.
LA GÉNÉTIQUE MOLÉCULAIRE DU SYNDROME DE KALLMANN
L’HH peut donc résulter d’une perturbation de l’action de la GnRH ou de sa sécrétion, d’anomalies dans les connections neuronales avec les neurones GnRH ou d’anomalies survenant au cours de la migration des neurones GnRH pendant le développement. Un élément clé permettant de différencier les anomalies probables du développement des anomalies fonctionnelles est la coexistence d’une anosmie/hyposmie.
Le syndrome de Kallmann est un déficit gonadotrope lié à une anomalie de migration des neurones GnRH et associé à une anosmie ou une hyposmie. À l’IRM, une aplasie ou une hypoplasie des bulbes olfactifs révèle le défaut de développement des bulbes olfactifs. Sa fréquence de 1/10 000 est probablement sous estimée. Le syndrome de Kallmann est dû à un défaut de développement des bulbes olfactifs entraînant un défaut de migration des neurones GnRH [43]. Trois modes de transmission sont décrits, lié au chromosome X, autosomique dominant et autosomique récessif.
Actuellement, huit gènes sont décrits dans le syndrome de Kallmann : des mutations inactivatrices sont retrouvées dans le gène KAL1 (anosmin-1, MIM : 308700) situé sur le chromosome X, ou dans des gènes situés sur des autosomes comme
FGFR1 (récepteur 1 du fibroblaste growth factor, MIM : 136350), PROK2 (prokinéticine 2
MIM : 607002),
PROKR2 (son récepteur MIM : 607212), FGF8 (fibroblaste growth factor 8 MIM : 600483),
NELF (nasal embryonic LHRH factor, MIM : 608137),
HS6ST1 (Heparan Sulfate, 6-O-sulfotransferase-1, MIM : 604846) ou WDR11 (WD repeat-containing protein 11, MIM : 606417). Ces gènes codent pour des protéines nécessaires au développement du bulbe olfactif [43-48]. Les mutations de FGFR1 représentent environ 10 % des cas de patients IHH et leur phénotype est très variable [49]. Cette variabilité de la profondeur du déficit gonadotrope et des signes associés permet d’évoquer, que le défaut de migration des neurones GnRH est parfois incomplet. Chez certains patients porteurs de mutations de FGFR1 , l’hypogonadisme est sévère, chez d’autres porteurs de la même mutation, le phénotype est réversible voir l’axe gonadotrope est normal [50]. Cette variabilité phénotypique dépend probablement de gènes modificateurs dont certains pourraient participer à l’activation du FGFR1 ou des facteurs de l’environnement.
Le syndrome CHARGE fait aussi partie des hypogonadismes hypogonadotropes associés à des anomalies de développement des neurones GnRH dues à une hypoplasie des bulbes olfactifs. Des mutations de CHD7 sont responsables du syndrome de charge. Des mutations de ce gène sont également décrites dans le syndrome de Kallmann.
Ces travaux de génétique moléculaire ont permis de revoir le mode de transmission de l’IHH. Sykiotis et collaborateurs ont mis en évidence en 2010 des variants rares de gènes d’IHH dans une cohorte de 397 patients caucasiens ayant un déficit en GnRH [24]. Ces variants étaient situés sur un seul allèle dans 17 % des cas chez les patients et 10 % des cas contrôles. Pour expliquer cette fréquence élevée de mutations chez les contrôles, les auteurs ont supposé que le HH était une maladie oligogénique. En effet, des variants rares bi-alléliques étaient retrouvée dans 2,5 % des cas des patients IHH dans deux ou plusieurs gènes. Par contre, aucun sujet contrôle n’était porteur de deux variants ou plus [24]. Ce résultat suggère un modèle oligogénique pour le déficit gonadotrope transmis selon un mode dominant ou récessif. Ils renforcent l’hypothèse de réseaux neuronaux participants à l’initiation de la puberté.
L’HYPOGONADISME HYPOGONADOTROPE SYNDROMIQUE
Le déficit gonadotrope est parfois dû à des mutations dans des gènes codant pour des protéines impliquées dans d’autres voies métaboliques, hormonales ou développementales. Elles sont alors responsables d’un hypogonadisme hypogonadotrope syndromique complexe [51].
L’inactivation de la leptine par exemple (LEP, MIM : 164160), de son récepteur (LEPR, MIM : 601007) ou plus exceptionnellement de la proconvertase 2 (PC2, MIM 600955) conduisent à un déficit gonadotrope associé à une obésité morbide (voir tableau 2). La proconvertase 2 est essentielle pour la maturation de plusieurs neuropeptides.
L‘inactivation de DAX1 (Dss — AHC — Critical Region on the X Chromosome,
Gene 1 ou NR0B1 ; MIM : 300473) associe déficit gonadotrope et hypoplasie des surrénales. C’est un gène impliqué dans la répression transcriptionnelle mais le mécanisme de l’HH associé à son déficit n’est pas connu.
D’autres gènes codent pour des protéines nécessaires à la différenciation des cellules gonadotropes hypophysaires. Leurs déficits associent un HH à d’autres déficits hypophysaires.
Le déficit gonadotrope est décrit également dans plusieurs syndromes dont les plus connus sont le syndrome de Prader-Willi et le syndrome de Bardet-Biedl. À la différence du syndrome de Kallmann, l’hypogonadisme est rarement le signe d’appel. Les mécanismes physiopathologiques du déficit gonadotrope sont généralement mal compris. La fréquence du syndrome de Prader-Willi est de 1/10 000-16 000. Ce syndrome est dû à une délétion en 15q11-q13 du chromosome paternel (70-75 % des cas), une disomie maternelle de la région 15q11-q13 (20-25 % des cas), une mutation du centre d’empreinte (2-5 % des cas), ou une translocation (1 % des cas). Le syndrome de Prader-Willi (SPW) associe un déficit intellectuel, une petite taille, une hypotonie, une obésité et des troubles du comportement [52]. Le déficit gonadotrope est d’intensité variable, et son mécanisme physiopathologique exact n’est pas connu mais pourrait être d’origine hypothalamique. Actuellement de nombreux travaux sont en cours pour mieux connaître l’implication des gènes SNRPN (small nuclear ribonucleoprotein polypeptide N) et Necdin dans le SPW, et dans les fonctions de l’hypothalamus notamment [53].
Le syndrome de Bardet-Biedl (prévalence de 1/125 000 à 1/175 000) associe une obésité, un déficit intellectuel, une polydactylie, une rétinite pigmentaire et un HH.
Le SBB est une ciliopathie, c’est-à-dire une affection liée à une atteinte des cils primitifs [54]. Chez l’homme, le SBB est génétiquement très hétérogène puisqu’il est la conséquence de mutations dans plusieurs gènes différents [55]. Le mécanisme du déficit gonadotrope est inconnu.
LA GÉNÉTIQUE DE LA PUBERTÉ PRÉCOCE CENTRALE
La puberté précoce centrale (PPC) peut être idiopathique. Il s’agit d’un diagnostic d’exclusion après avoir éliminé les causes organiques et notamment tumorale. La PPC est parfois familiale avec plusieurs cas de PPC au sein de la famille. Il s’agit d’une pathologie plus fréquente chez fille. Un terrain génétique favorable est souvent évoqué devant une avance pubertaire notamment chez la mère du propositus. La PPC est actuellement considérée comme une puberté normale qui survient plus tôt.
La fonction de reproduction est généralement normale à l’âge adulte. Le traitement est justifié devant le risque de petite taille par arrêt de la croissance prématurée.
Les récepteurs KISS1R, TAC3R et ProKR2 sont des récepteurs couplés aux protéines G, famille de récepteurs comprenant de nombreux exemples de mutations constitutives responsables d’activation en absence de ligand. Récemment, une mutation de KiSS1R , a été rapportée dans un cas de puberté précoce centrale [56].
Cette mutation prolonge l’activation du récepteur par les kisspeptins. Il s’agit d’une hypothèse intéressante qui soulève de nombreuses questions non résolues telle que la persistance de la pulsatilité de la sécrétion de la GnRH chez cette enfant. KISS1 était également un gène candidat intéressant pour la puberté précoce centrale.
Récemment, nous avons montré l’association entre certains polymorphismes de la région 3′non traduite de KISS1 et la puberté précoce centrale [57]. Nous montrons que ces polymorphismes perturbent la conformation de l’ADN en G-quadruplex dont la fonction serait la fixation de protéines régulatrices de la stabilité des ARNm.
Ce résultat suggère une anomalie de la régulation post-transcriptionnelle de KISS1 dans la puberté précoce centrale.
CONCLUSION
Les avancées les plus importantes de ces dernières années dans la compréhension de l’initiation de la puberté ont été obtenues grâce à l’étude de la génétique de la puberté normale et des maladies de l’initiation de la puberté. Ces deux approches complémentaires confirment que l’initiation de la puberté résulte d’une maturation post-natale de l’hypothalamus dont le but ultime est l’augmentation de la sécrétion de la GnRH par les neurones de l’hypothalamus. Il est maintenant possible de définir un réseau de neurones hypothalamiques participant à cette régulation. De nombreuses boucles autocrines et paracrines sont évoquées à partir de modèles animaux. La chronologie de l’activation de ce réseau détermine l’âge de la puberté.
Cette activation programmée dans le temps semble dépendre de gènes dits hétérochoniques tel que Lin28b qui code pour une protéine qui participe à la régulation de la maturation de petits ARN non codants. La poursuite de cette approche de génétique humaine devrait permettre de mieux caractériser ce réseau hypothalamique et notamment définir les mécanismes neuronaux qui participent à cette maturation.
BIBLIOGRAPHIE [1] Parent AS, Teilmann G, Juul A, et al. — The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration.
Endocr. Rev ., 2003, 24, 668-693.
[2] Ebling FJ. — The neuroendocrine timing of puberty.
Reproduction , 2005, 129 , 675-683.
[3] Villanueva C, de Roux N. — [Biological mechanisms and genes involved in puberty].
Rev.
Prat ., 2008, 58 , 1305-1309.
[4] Conte FA, Grumbach MM, Kaplan SL, Reiter EO. — Correlation of luteinizing hormonereleasing factor-induced luteinizing hormone and follicle-stimulating hormone release from infancy to 19 years with the changing pattern of gonadotropin secretion in agonadal patients:
relation to the restraint of puberty. The Journal of clinical endocrinology and metabolism , 1980, 50 , 163-168.
[5] Plant TM WS. —
Puberty in Nonhuman Primates and Human New York: Raven Press, 2007.
[6] Sisk CL, Foster DL. — The neural basis of puberty and adolescence.
Nat. Neurosci ., 2004, 7 , 1040-1047.
[7] Towne B, Czerwinski SA, Demerath EW, et al. — Heritability of age at menarche in girls from the Fels Longitudinal Study.
Am. J. Phys. Anthropol ., 2005, 128 , 210-219.
[8] Ong KK, Elks CE, Li S, et al. — Genetic variation in LIN28B is associated with the timing of puberty.
Nat. Genet ., 2009.
[9] Sulem P, Gudbjartsson DF, Rafnar T, et al. — Genome-wide association study identifies sequence variants on 6q21 associated with age at menarche.
Nat. Genet ., 2009.
[10] He C, Kraft P, Chen C, et al. — Genome-wide association studies identify loci associated with age at menarche and age at natural menopause.
Nat. Genet ., 2009.
[11] Perry JR, Stolk L, Franceschini N, et al. — Meta-analysis of genome-wide association data identifies two loci influencing age at menarche.
Nat. Genet ., 2009.
[12] Viswanathan SR, Daley GQ, Gregory RI. — Selective blockade of microRNA processing by Lin28. Science , 2008, 320 , 97-100.
[13] Krstevska-Konstantinova M, Charlier C, Craen M, et al. — Sexual precocity after immigration from developing countries to Belgium: evidence of previous exposure to organochlorine pesticides. Hum. Reprod ., 2001, 16 , 1020-1026.
[14] Teilmann G, Pedersen CB, Skakkebaek NE, Jensen TK. — Increased risk of precocious puberty in internationally adopted children in Denmark. Pediatrics , 2006, 118 , e391-399.
[15] INSERM, Centre CE.
Aspects génétiques de la puberté Paris: Editions INSERM, 2000-2007.
[16] Mason AJ, Hayflick JS, Zoeller RT, et al. — A deletion truncating the gonadotropinreleasing hormone gene is responsible for hypogonadism in the hpg mouse.
Science , 1986, 234 , 1366-1371.
[17] Bouligand J, Guiochon-Mantel A, Young J. — [GNRH1 mutation in familial hypogonadotropic hypogonadism]. Medecine sciences : M/S . 2009, 25 , 791-793.
[18] Chan YM, de Guillebon A, Lang-Muritano M, et al. — GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism.
Proceedings of the National Academy of Sciences of the United States of America , 2009, 106 , 11703-11708.
[19] Chevrier L, Guimiot F, de Roux N. — GnRH receptor mutations in isolated gonadotropic deficiency. Molecular and cellular endocrinology , 2011.
[20] de Roux N, Young J, Misrahi M, et al. — A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor.
The New England journal of medicine , 1997, 337 , 1597-1602.
[21] Layman LC, Cohen DP, Jin M, et al. — Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism.
Nature genetics , 1998, 18 , 14-15.
[22] de Roux N, Young J, Brailly-Tabard S, et al. — The same molecular defects of the gonadotropin-releasing hormone receptor determine a variable degree of hypogonadism in affected kindred. The Journal of clinical endocrinology and metabolism , 1999, 84 , 567-572.
[23] Topaloglu AK, Lu ZL, Farooqi IS, et al. — Molecular genetic analysis of normosmic hypogonadotropic hypogonadism in a Turkish population: identification and detailed functional characterization of a novel mutation in the gonadotropin-releasing hormone receptor gene.
Neuroendocrinology , 2006, 84 , 301-308.
[24] Sykiotis GP, Plummer L, Hughes VA, et al. — Oligogenic basis of isolated gonadotropinreleasing hormone deficiency.
Proceedings of the National Academy of Sciences of the United States of America , 2010, 107 , 15140-15144.
[25] de Roux N, Genin E, Carel JC, et al. — Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54.
Proc. Natl. Acad. Sci. U S A , 2003, 100 , 10972-10976.
[26] Seminara SB, Messager S, Chatzidaki EE, et al. — The GPR54 gene as a regulator of puberty.
The New England journal of medicine , 2003, 349 , 1614-1627.
[27] Lanfranco F, Gromoll J, von Eckardstein S, et al. — Role of sequence variations of the
GnRH receptor and G protein-coupled receptor 54 gene in male idiopathic hypogonadotropic hypogonadism. Eur. J. Endocrinol ., 2005, 153 , 845-852.
[28] Semple RK, Achermann JC, Ellery J, et al. — Two novel missense mutations in g proteincoupled receptor 54 in a patient with hypogonadotropic hypogonadism.
The Journal of clinical endocrinology and metabolism , 2005, 90 , 1849-1855.
[29] Tenenbaum-Rakover Y, Commenges-Ducos M, Iovane A, et al. — Neuroendocrine phenotype analysis in five patients with isolated hypogonadotropic hypogonadism due to a L102P inactivating mutation of GPR54. The Journal of clinical endocrinology and metabolism , 2007, 92 , 1137-1144.
[30] Teles MG, Trarbach EB, Noel SD, et al. — A novel homozygous splice acceptor site mutation of KISS1R in two siblings with normosmic isolated hypogonadotropic hypogonadism. Eur. J. Endocrinol ., 2010, 163 , 29-34.
[31] Nimri R, Lebenthal Y, Lazar L, et al. — A novel loss-of-function mutation in
GPR54/KISS1R leads to hypogonadotropic hypogonadism in a highly consanguineous family.
The Journal of clinical endocrinology and metabolism , 2011, 96 , E536-545.
[32] Oakley AE, Clifton DK, Steiner RA. — Kisspeptin signaling in the brain.
Endocr. Rev. , 2009, 30 , 713-743.
[33] Ojeda SR UH. —
Puberty in the rat New York: Raven Press, 2007.
[34] Topaloglu AK, Tello JA, Kotan LD, et al. — Inactivating KISS1 mutation and hypogonadotropic hypogonadism.
N. Engl. J. Med ., 2012, 366 , 629-635.
[35] Topaloglu AK, Reimann F, Guclu M, et al. — TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat. Genet ., 2009, 41 , 354-358.
[36] Karges B, Neulen J, de Roux N, Karges W. — Genetics of isolated hypogonadotropic hypogonadism: role of GnRH receptor and other genes. International journal of endocrinology , 2012, 2012 , 147893.
[37] Ramaswamy S, Seminara SB, Ali B, et al. — Neurokinin B stimulates GnRH release in the male monkey (Macaca mulatta) and is colocalized with kisspeptin in the arcuate nucleus.
Endocrinology , 2010, 151 , 4494-4503.
[38] Billings HJ, Connors JM, Altman SN, et al. — Neurokinin B acts via the neurokinin-3 receptor in the retrochiasmatic area to stimulate luteinizing hormone secretion in sheep.
Endocrinology , 2010, 151 , 3836-3846.
[39] Navarro VM, Castellano JM, McConkey SM, et al. — Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat.
American journal of physiology Endocrinology and metabolism , 2011, 300 , E202-210.
[40] Kung TT, Crawley Y, Jones H, et al. — Tachykinin NK3-receptor deficiency does not inhibit pulmonary eosinophilia in allergic mice.
Pharmacol. Res ., 2004, 50 , 611-615.
[41] Funes S, Hedrick JA, Vassileva G, et al. — The KiSS-1 receptor GPR54 is essential for the development of the murine reproductive system.
Biochemical and biophysical research communications , 2003, 312 , 1357-1363.
[42] Chan YM, Broder-Fingert S, Wong KM, Seminara SB. — Kisspeptin/Gpr54-independent gonadotrophin-releasing hormone activity in Kiss1 and Gpr54 mutant mice. Journal of neuroendocrinology , 2009, 21 , 1015-1023.
[43] Dode C, Levilliers J, Dupont JM, et al. — Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome.
Nat. Genet ., 2003, 33 , 463-465.
[44] Legouis R, Hardelin JP, Levilliers J, et al. — The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules.
Cell ., 1991, 67 , 423-435.
[45] Dode C, Teixeira L, Levilliers J, et al. — Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2.
PLoS Genet ., 2006, 2 , e175.
[46] Falardeau J, Chung WC, Beenken A, et al. — Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice.
J. Clin. Invest ., 2008, 118 , 2822-2831.
[47] Miura K, Acierno JS, Jr., Seminara SB. — Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH). Journal of human genetics , 2004, 49 , 265-268.
[48] Kim HG, Ahn JW, Kurth I, et al. — WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. American journal of human genetics , 2010, 87 , 465-479.
[49] Villanueva C, de Roux N. — FGFR1 mutations in Kallmann syndrome.
Front Horm. Res ., 2010, 39 , 51-61.
[50] Pitteloud N, Acierno JS, Jr., Meysing AU, et al. — Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. The Journal of clinical endocrinology and metabolism , 2005, 90 , 1317-1322.
[51] Roze C, Touraine P, Leger J, de Roux N. — [Congenital hypogonadotropic hypogonadism].
Ann. Endocrinol. (Paris) , 2009, 70 , 2-13.
[52] Prader A. — [Adrenogenital syndrome, adrenogenital salt deficiency syndrome and Cushing’s syndrome in childhood]. Schweiz Med. Wochenschr ., 1956, 86 , 289-299.
[53] Miller NL, Wevrick R, Mellon PL. — Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum. Mol. Genet ., 2009, 18 , 248-260.
[54] Bisgrove BW, Yost HJ. — The roles of cilia in developmental disorders and disease.
Develo- pment , 2006, 133 , 4131-4143.
[55] Laurier V, Stoetzel C, Muller J, et al. — Pitfalls of homozygosity mapping: an extended consanguineous Bardet-Biedl syndrome family with two mutant genes (BBS2, BBS10), three mutations, but no triallelism. Eur. J. Hum. Genet ., 2006, 14 , 1195-1203.
[56] Teles MG, Bianco SD, Brito VN, et al. — A GPR54-activating mutation in a patient with central precocious puberty.
N. Engl. J. Med ., 2008, 358 , 709-715.
[57] Huijbregts L, Roze C, Bonafe G, et al. — DNA polymorphisms of the KiSS1 3′untranslated region interfere with the folding of a G-rich sequence into G-quadruplex.
Mol. Cell Endocrinol ., 2012, 351 , 239-248.
DISCUSSION
M. Philippe BOUCHARD
Le peptide Kiss qui stimule les neurones à GnRH est-il impliqué dans le fonctionnement pulsatile de ceux-ci (intermittents) ? Kiss déclenche la puberté, quel(s) facteur(s) déclenche(nt) le fonctionnement des neurones Kiss, à la puberté ?
L’analyse du phénotype neuroendocrinien d’une patiente ayant une aménorrhée primaire due à deux mutations hétérozygotes composites du récepteur KISS1R montre la persistance d’une pulsatilité de la LH avec des pics de faible amplitude chez cette patiente.
Ce résultat montre que l’inactivation de KISS1R ne perturbe pas la pulsatilité intrinsè- que de la GnRH. Ce schéma a depuis été confirmé par plusieurs travaux expérimentaux chez les rongeurs, à partir d’explants hypothalamiques ou de cultures cellulaires. Le groupe de Terasawa (USA) propose une sécrétion pulsatile des kisspeptins ce qui pourrait expliquer en partie la pulsatilité de la GnRH. Les facteurs déclenchant l’activation des neurones kisspeptin à la puberté ne sont pas connus. Le démarrage de la puberté est un mécanisme progressif. Il est supposé un mécanisme amplificateur entre les neurones Kisspeptins et les hormones sexuelles par l’intermédiaire du rétrocontrole positif de l’estradiol sur l’expression des kisspeptins dans le noyau AVPV.
M. René MORNEX
Après avoir présenté le rôle ambivalent du Kiss-peptide à la fois stimulant et freinateur, votre schéma terminal du rôle du Kiss dans le déclenchement de la puberté ne retient que l’effet +. N’y a-t-il pas de mobilisation de l’effet freinateur ?
Il n’existe pas, à ce jour, d’argument en faveur d’un effet freinateur des kisspeptins sur l’axe gonadotrope, les kisspeptins sont des neuropeptides activateurs de la sécrétion de la GnRH. Les travaux chez la souris et le rat montrent une augmentation de l’expression des Kisspeptins dans l’AVPV avant la puberté mais pas de différence nette de l’expression dans le noyau arqué. La gonadectomie chez l’adulte entraine une augmentation de l’expression de KISS1 dans le noyau arqué et une baisse de l’expression dans l’AVPV alors que la gonadectomie chez la souris pré-pubère empêche l’augmentation de l’expres- sion de
KISS1 dans l’AVPV. Il est vrai que cette observation est étonnante. Elle indique que le tonus de base des kisspeptins sur l’axe gonadotrope dépend essentiellement du noyau arqué chez l’adulte. Par contre, chez la souris pré-pubère, les kisspeptin exprimés dans les neurones de l’AVPV sont déterminants pour initier la puberté à l’âge normal.
M. Edwin MILGROM
Dans les hypogonadismes hypogonadotropes isolées congénitales, lorsque vous séquencez les différents gènes impliqués jusqu’ici, dans quel pourcentage des cas le résultat reste-t-il négatif ? Autrement dit reste-t-il d’autres gènes à découvrir ?
Nos travaux montrent clairement qu’il existe d’autres gènes à découvrir. Les arguments bien sur la fréquence relativement faible, environ 10 % de mutations dans les gènes connus pour l’ensemble des hypogonadismes hypogonadotropes isolés. Lorsque l’analyse est restreinte aux formes familiales d’IHH avec transmission récessive, la fréquence de mutations dans les gènes connus est supérieure à 60 %. Nous avons maintenant la certitude que certaines familles avec transmission récessive ne sont pas liées aux loci des gènes connus ce qui permet d’affirmer l’existence d’autres gènes. Ces nouveaux gènes sont en cours de caractérisation par plusieurs équipes.
M. Claude JAFFIOL
Quel est le lien entre les facteurs nutritionnels et la régulation hypothalamique de la puberté ? Quelle est l’interférence avec les Kiss-peptides ?
Le lien entre nutrition et puberté est connu depuis de nombreuses années. Les états de dénutrition sont responsables de retard pubertaire. Sur le plan de la régulation hypothalamique, de nombreux neuropeptides participant à la régulation de la prise alimentaire participent également à la régulation de l’axe gonadotrope. Le plus connu étant la leptine bien que des travaux récents suggèrent que le déficit gonadotrope observé dans les cas de mutation de la leptine ou de son récepteur ne serait pas du à la perte de l’activation par la leptine des neurones hypothalamiques de l’axe gonadotrope. On peut également citer le NPY, les mélanocortines, l’alpha MSH, la Ghrelin. Le lien entre facteurs nutritionnels et kisspeptin a été étudié essentiellement à partir de modèles de rongeurs. Il apparait que les kisspeptin n’ont pas d’effet sur la prise alimentaire. Par contre, les conditions métaboliques entraînant un déficit gonadotrope ou un retard pubertaire diminuent l’expression des kisspeptins dans l’hypothalamus. L’administration de kisspeptins chez ces animaux réactive l’axe gonadotrope ce qui confirme le déficit fonctionnel en kisspeptins dans ces états de dénutrition. A l’opposé, dans certains modèles d’obésité chez le rongeur, il a été observé une diminution de l’expression des kisspeptins. Le lien entre neuropeptides hypothalamiques impliqués dans la prise alimentaire et kisspeptins a été largement étudié. Il est difficile de résumer simplement ces différents travaux. Ils montrent néanmoins clairement un lien étroit entre facteurs métaboliques et kisspeptins.
M. Christian NEZELOF
La puberté s’accompagne souvent chez le garçon d’une gynécomastie. Quelle est son origine ?
La gynécomastie du garçon pendant la puberté est due à une augmentation de la sensibilité aux estrogènes des cellules mammaires dans une situation de relatif déficit en androgènes en début de puberté.
M. Pierre JOUANNET
Les polymorphismes de Kiss sont-ils aussi fréquents dans les deux sexes ? Quelle est la fertilité des personnes porteuses de polymorphismes de Kiss ? Serait-il légitime de chercher des polymorphismes de Kiss chez les garçons atteints de cryporchidie à la naissance ?
Il paraît peu probable que la fréquence des polymorphismes de
KISS1 , soit différente entre les deux sexes. Nous n’avons pas connaissance d’études sur l’association entre polymorphismes de KISS1 et fertilité. La recherche de polymorphismes de KISS1 chez les enfants ayant une cryptorchidie, pourrait être justifiée. Nos résultats montrent que le système kisspeptin participe à la régulation de l’activation de l’axe gonadotrope durant la vie fœtale. Néanmoins, cette hypothèse suppose un polymorphisme qui modifie la synthèse des kisspeptins provoquant un défaut de migration testiculaire sans entraîner de défaut de développement du pénis qui dépend également des androgènes.
M. Jacques BATTIN
La maturation progressive du gonadostat féminin est attestée par les cycles anovulatoires de la période postménarchiale d’infertilité relative. A-t-on des données sur la maturation neuro-génétique des initiateurs de la puberté ?
Il s’agit d’une question complexe Les cycles anovulatoires de la période postnémarchiale sont notamment en rapport avec des pics ovulatoires de LH de mauvaise qualité. Il est maintenant admis que le pic ovulatoire de LH dépend des kisspeptins par un mécanisme de rétro-contrôle positif de l’œstradiol sur les neurones kisspeptins. Il existe donc un lien entre initiateurs de la puberté et régulation de l’ovulation. L’expression des kisspeptins dépendant des estrogènes, on peut donc supposer que les cycles anovulatoires sont dûs à un défaut de synthèse de kisspeptins par immaturité du rétro-contrôle positif. C’est probablement la situation qui prévaut dans la période postménarchiale. La notion de maturation neuro-génétique que vous proposez suggère la participation de facteurs épigénétiques qui moduleraient l’expression des gènes dont notamment KISS1 . Il n’existe pas de travaux à notre connaissance en faveur de cette intéressante hypothèse.
M. Emmanuel-Alain CABANIS
En matière d’hypogodanisme secondaire, quelle est la proportion de processus occupant l’espace (gliome opto-chiasmatique, cranio-pharygiome…), chiasmato-hypophysaire, au sein du tableau que vous nous avez présenté ? Vous avez évoqué l’absence de migration neuronale (à relier aux dystrophysmes associés que vous avez cités), mais qu’en est-il de la myélinisation, à travers des maladies dysmyélinisantes congénitales, des retards de myélinisation ou des maladies inflammatoires démyélinisantes du système nerveux central ?
Il est certain qu’il s’agit d’une question fondamentale qui recentre le débat sur la régulation neuronale et pas seulement peptidique de l’axe gonadotrope et du processus pubertaire. Les neurotransmetteurs inhibiteurs ou activateurs sont largement impliqués dans la régulation de la sécrétion de la GnRH. Le rôle de la plasticité synaptique dans l’initiation de la puberté a notamment été évoqué par de nombreux groupes. La question de la myélinisation commence à être abordée notamment par l’étude du 4H syndrome qui comprend hypomyélinisation, hypodontie et hypogonadisme hypogonadotrope. Ce syndrome est du à des mutations de sous unités de la RNA polymérase III. La compréhension de cette association pourrait ouvrir des voies de recherche très intéressantes pour comprendre l’initiation de la puberté.
Bull. Acad. Natle Méd., 2012, 196, no 2, 327-343, séance du 14 février 2012