
Résumé
Les mécanismes épigénétiques jouent un rôle clé dans la régulation de l’expression des gènes. Une des modifications épigénétiques les plus étudiées est la méthylation au niveau des dinucléotides CpG que ce soit au niveau des promoteurs des gènes, des transposons ou des centres de contrôle de l’empreinte (ICR). L’empreinte génomique est une marque épigéné- tique qui est dépendante de l’origine parentale et qui est associée à une expression génique monoallèlique. Plusieurs gènes impliqués dans la croissance embryonnaire et fœtale sont soumis à l’empreinte génomique. Il existe deux périodes critiques dans la mise en place ou le maintien de l’empreinte : lors de la gamétogénèse et lors du développement précoce préimplantatoire du zygote. La reprogrammation majeure prend place dans les cellules germinales primordiales dans lesquelles l’empreinte est effacée et où leur totipotence est restaurée. Les marques de l’empreinte sont ensuite apposées durant la spermatogénèse ou l’ovogénèse en fonction du sexe. Après la fertilisation, il existe une déméthylation globale du génome qui sera suivie d’une vague de méthylation de novo, mais dont les loci soumis à l’empreinte sont épargnés. Des anomalies de l’empreinte peuvent entraîner des troubles de la croissance fœtale comme observés dans le syndrome de croissance excessive de BeckwithWiedemann (BWS), ou au contraire le retard de croissance intra utérin du syndrome de Silver-Russell (RSS).Ces syndromes sont dûs à des méthylations d’ADN anormales au niveau de la région chromosomique 11p15 où sont localisés de nombreux gènes soumis à l’empreinte, dont le facteur de croissance IGF2. Dans les BWS une perte de méthylation au niveau de cette région centromérique ICR2 (KCN1OT1) de l’allèle maternel ou un gain de méthylation de la région télomérique ICR1/IGF2/H19 sur l’allèle maternel ont été montrés ; cette dernière anomalie étant associée à un risque élevé de tumeur pédiatrique dont le néphroblastome. À l’opposé une déméthylation d’ICR1 de l’allèle paternel a été montré dans le miroir clinique qu’est le RSS. La gamétogénèse et la période précoce post fertilisation représentent des fenêtres critiques de perturbation de la mise en place de l’empreinte par des facteurs environnementaux. Une fréquence anormale d’aide médicale à la procréation a été notée dans les séries de BWS suggérant que celle-ci puisse avoir une incidence sur les anomalies de l’empreinte. Les causes à l’origine de ces anomalies d’empreinte ne sont pas connues. De plus d’autres anomalies de l’empreinte touchant de nombreux loci autre que ceux de la région 11p15 ont été décrites que ce soit chez des enfants issus d’AMP ou de grossesse spontanée. Ces données suggèrent que les troubles de la méthylation de l’ADN survenant après la fécondation implique un (des) facteur contrôlant en trans la régulation des marques de méthylation.
Summary
Epigenetic phenomena play a key role in regulating gene expression. One of the most widely studied epigenetic modification is DNA methylation at cytosine residues of CpG dinucleotides in gene promoters, transposons and imprinting control regions (ICR). Genomic imprinting refers to epigenetic marking of genes that results in monoallelic expression depending on the parental origin. Several genes encoding key hormones involved in embryonic and fetal growth are imprinted. There are two critical periods of epigenetic reprogramming: gametogenesis and early preimplantation development. Major reprogramming takes place in primordial germ cells, in which parental imprints are erased and totipotency is restored. Imprint marks are then re-established during spermatogenesis or oogenesis, depending on gender. Upon fertilization, genome-wide demethylation is followed by a wave of de novo methylation, both processes being resisted by imprinted loci. Disruption of imprinting can cause growth defects such as the Beckwith-Wiedemann overgrowth syndrome (BWS) and the Russell-Silver (RSS) intrauterine and postnatal growth retardation syndrome. These growth disorders are caused by abnormal DNA methylation in the 11p15 imprinted region encompassing many imprinted genes, such as IGF2. BWS has been linked to loss of methylation (LOM) in the centromeric ICR2/KCNQ1OT1 region of the maternal allele, or gain of methylation in the telomeric ICR1/IGF2/H19 region of the maternal allele. This latter epigenetic defect is associated with an increased risk of tumors such as nephroblastoma. LOM in the telomeric ICR1 region of the paternal allele has been detected in RSS. Early embryogenesis is a critical period of epigenetic regulation, and is sensitive to environmental factors. Individuals conceived with the help of assisted reproductive technology (ART) are over-represented among BWS patients, suggesting that ART may favor altered imprinting at the imprinted centromeric 11p15 locus (LOM in the maternally methylated ICR2 region). The underlying cause of these imprinting defects, both spontaneous and ART-related, is unclear. However, recent data show that, in patients with BWS or RSS, including those conceived with the help of ART, the DNA methylation defect involves imprinted loci other than 11p15. This suggests that unfaithful maintenance of DNA methylation marks following fertilization involves dysregulation of a trans-acting regulatory factor.
INTRODUCTION
L’anomalie d’expression d’un gène, l’absence de synthèse de la protéine correspondante ou la synthèse d’une protéine inactive sont les conséquences classiques d’une anomalie génétique. Il peut s’agir d’une mutation nucléotidique, d’une délétion dans la zone codante, d’un défaut de contrôle de la transcription par anomalie des séquences promotrices et régulatrices ou enfin de déficit en molécules s’associant aux promoteurs de ces gènes. Si le code génétique est connu depuis longtemps, ce n’est que ces vingt dernières années que les phénomènes épigénétiques contrôlant l’expression des gènes ont été décrits. Ceux-ci sont impliqués dans des processus physiologiques du développement et de la différenciation cellulaire (expliquant qu’une cellule différenciée ait perdu sa totipotence en s’engageant progressivement dans des processus en principe irréversibles), ou pathologiques comme les processus tumoraux.
Épigénétique et empreinte génomique
La régulation de l’expression des gènes n’est pas régulée uniquement par le code génétique mais aussi par des modifications épigénétiques. L’empreinte parentale est une modification épigénétique particulière car elle touche uniquement un allèle et induit une différence de fonction entre deux allèles de séquence nucléotidique identique.
Le contrôle épigénétique est représenté par l’ensemble des modifications non contenues dans la séquence nucléotidique de l’ADN (code génétique) mais qui sont cependant héritables, c’est-à-dire transmissibles lors de la mitose, et qui contrôlent l’expression des gènes. L’unité de base de l’organisation chromatinienne est repré- sentée par le nucléosome constitué d’un octamère de protéines histones autour duquel s’enroule le brin d’ADN (Fig. 1). La marque épigénétique la mieux connue est la méthylation de l’ADN au niveau d’îlots CpG (domaines d’ADN très riches en dinucléotides CG). Les modifications post-traductionnelles (acétylation, méthylation…) des histones H3 et H4 ont aussi un rôle déterminant dans la régulation épigénétique. Au niveau d’une région chromatinienne, les différentes modifications post-traductionnelles des histones (code histone) et de l’ADN (méthylé/non méthylé) vont induire soit la compaction de la chromatine (l’expression génique est alors réprimée), soit la décondensation de la chromatine (la régulation de la transcription est alors possible du fait de l’accessibilité des facteurs stimulateurs ou répresseurs aux régions régulatrices des gènes) [1].
De façon générale, la méthylation de l’ADN est associée à une dé-acétylation des histones dans les régions où la chromatine est compactée empêchant l’expression génique. Au contraire, lorsque l’ADN est déméthylé et les histones acétylées, la chromatine est dans une conformation ouverte permettant l’expression des gènes (Fig. 1).
L’empreinte parentale ou empreinte génomique a été mise en évidence chez les mammifères dans les années 80 grâce à des expériences de transferts nucléaires qui ont permis de démontrer la non équivalence des deux génomes parentaux :
la formation de zygotes gynogénotes (deux génomes maternels) conduit à un développement embryonnaire mais à l’absence de développement des annexes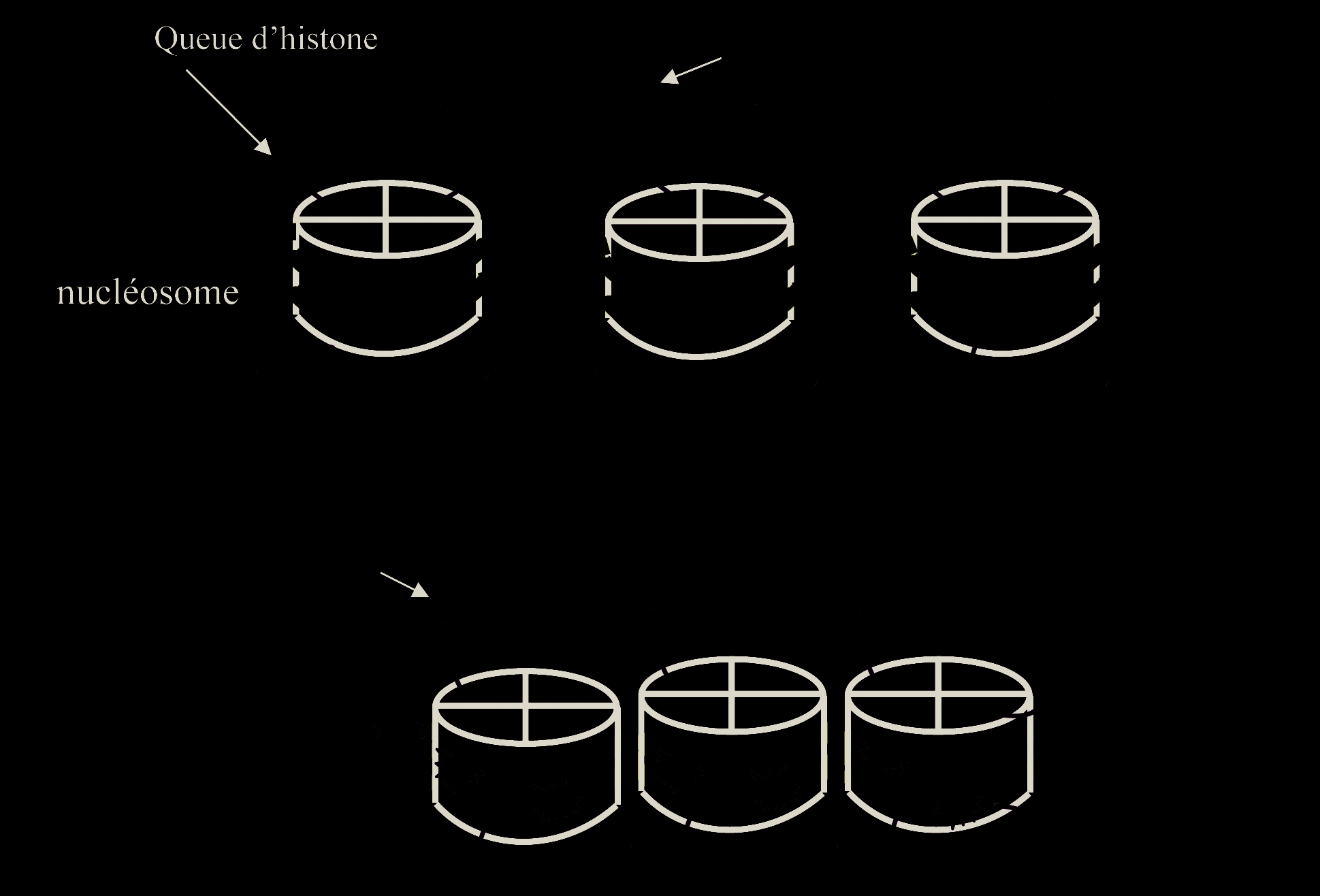
Fig. 1. — Exemples de marques épigénétiques De façon générale lorsque les queues d’histones sont acétylées et l’ADN non méthylé au niveau des ilots CpG, la chromatine est sous forme non compacte et la régulation de la transcription possible.
Dans le cas contraire la méthylation de l’ADN, la dé-acétylation des queues histones et leur méthylation sont associées à la compaction de la chromatine et à l’absence de régulation de l’expression.
H3 : histone 3, H4 : histone 4, K9 et K27 : Lysine 9 et 27 des queues d’histone.
embryonnaires alors que la formation de zygotes androgénotes (deux génomes paternels) conduit au développement des annexes sans développement embryonnaire. Ces expériences ont démontré que pour obtenir un développement normal, il ne suffisait pas d’avoir un génome diploïde mais qu’une contribution paternelle et maternelle était indispensable, faisant suspecter l’expression allèle spécifique de gènes impliqués dans le développement précoce [2]. L’empreinte parentale était ainsi mise en évidence. L’empreinte parentale dite différentielle encore appelée empreinte génomique, est un des aspects de la régulation épigénétique. Elle a été décrite d’abord pour un gène précis grâce à l’inactivation (« knock out ») du gène d’IGF-2 chez la souris [3]. Les souris hétérozygotes pour la mutation du gène d’IGF-2 présentaient un retard de croissance à la naissance uniquement quand la mutation provenait de l’allèle d’origine paternelle. La croissance était normale en cas de transmission maternelle du gène muté. Les souris homozygotes pour la mutation ne présentaient pas de phénotype plus sévère que les souris hétérozygotes avec mutation de l’allèle paternel. Alors que les deux loci sont identiques quant à leur séquence nucléotidique (code génétique), ils sont donc fonctionnellement différents. L’expression est monoallélique et un seul des deux allèles parentaux est
Fig. 2A. — La région 11p15 est soumise à l’empreinte parentale et est divisée en deux domaines, centromérique et télomérique. L’empreinte réciproque du gène H19 (ARN non codant) exprimée par l’allèle maternel (M) et le gène
IGF-2 exprimé par l’allèle paternel (P) dépend de l’ ICR1 méthylé différentiellement en amont du gène H19 . Cet ICR1 agit ainsi comme un insulateur. Le facteur CTCF lie l’
ICR1 maternel non méthylé et empêche le promoteur du gène d’ IGF-2 d’interagir avec les « enhancers » se trouvant en aval du gène H19 . Ceci résulte en un silence transcriptionnel de l’allèle
IGF-2 maternel. Sur l’allèle paternel ICR1 est méthylé prévenant la liaison de CTCF, et conduit à la transcription d’
IGF-2 paternel. Le domaine centromérique ICR2 fonctionne comme un « silenceur » en produisant un ARN non codant (l’ARN KCNQ1OT1 ). Ceci induit le silence de tous les gènes paternels du domaine dont CDKN1C (inhibiteur des cyclines cdk de la phase G1 et donc du cycle cellulaire). L’ARN KCNQ1OT1 est probablement impliqué dans les modifications d’histones des gènes voisins. Les gènes paternels exprimés sont représentés par les boîtes bleues, les gènes maternels par les boîtes oranges et les non-exprimés par les boîtes grises.
Fig. 2B. — Dans le cas du syndrome de Silver-Russell (RSS) la déméthylation anormale d’ICR1 au niveau de l’allèle paternel entraîne l’expression biallèlique d’H19 et inhibe l’expression d’IGF-2, responsable du retard de croissance intra-utérin.
Fig. 2C. — Dans le cas du syndrome de Beckwith Wiedemann (BWS), un des mécanismes pathologiques est dû à la méthylation anormale d’ICR1 au niveau de l’allèle maternel entraînant l’expression biallèlique d’IGF-2, responsable de la macrosomie, et l’inhibition d’expression de H19.
exprimé tandis que l’autre allèle reste silencieux. Alors que la grande majorité des gènes sont exprimés par les deux allèles parentaux, moins de 1 % des gènes sont soumis à empreinte parentale. Ces deux allèles ont subi des modifications épigéné- tiques différentes (méthylation, acétylation….) selon leur origine parentale aboutissant à l’expression ou à la non expression d’un gène en fonction de cette origine parentale (Fig 1). Ces marques épigénétiques sont effacées dans les gamètes, puis apposées de façon différente en fonction du sexe de l’individu, lors de la maturation des gamètes. Après la fécondation, durant la période préimplantatoire, ces marques épigénétiques doivent être protégées d’une vague de déméthylation puis d’une reméthylation globale du génome [1]. Au cours de ces dernières années, plusieurs régions chromosomiques soumises à empreinte et donc fonctionnellement différentes en fonction de l’origine parentale, ont été décrites. Les gènes soumis à empreinte sont regroupés en plusieurs « clusters » sur le génome (6q24-27, 7q31-32, 11p15, 15q11-13….) suggérant un contrôle coordonné des gènes d’une région soumise à l’empreinte. Certaines régions méthylées différentiellement (appelées DMR) repré- sentent les centres d’empreinte (Imprinting Center Region : ICR) qui régulent l’expression monoallélique de plusieurs gènes dans une région donnée.
À côté des méthyltransférases d’ADN et des enzymes modifiant l’acétylation et la méthylation des histones, il existe d’autres protéines régulatrices de l’expression génique notamment les protéines qui se lient aux ilots CpG en fonction de leur état de méthylation et notamment des protéines « insulatrices » capables, comme le facteur CTCF, de constituer une « barrière » entre un « enhancer » et les promoteurs de plusieurs gènes du domaine (cf exemple H19, IGF-2 : Fig 2). De plus certains gènes soumis à empreinte codent pour des ARN non traduits (H19, KCNQ1OT1) dont on ne connaît pas encore parfaitement le rôle. Ils inhiberaient l’expression des gènes adjacents.
Système IGF et empreinte
Chez les mammifères, le système des insulin-like growth factors ou IGFs comporte deux ligands structurellement homologues (IGF-1 et IGF-2), six protéines de liaison (insulin-like growth factor-binding proteins : IGFBPs 1-6) et deux récepteurs. La signalisation de ces deux IGF est assurée par le récepteur IGF-1R.
IGF-2R, quant à lui, assure la clairance d’IGF-2. Le système des IGF est largement impliqué dans le métabolisme intermédiaire, la prolifération, la différenciation et la survie cellulaires, le développement et leurs anomalies sont responsables de nombreuses pathologies de la croissance fœtale, post natale et tumorale. L’inactivation des gènes du système IGF [3, 4] a notamment montré non seulement l’importance d’IGF2 dans le développement et la croissance du fœtus, mais aussi qu’il était soumis à l’empreinte génomique. Il est exprimé dès les premiers stades embryonnaires de façon ubiquiste et sa concentration sérique est très élevée durant toute la période fœtale.
Pathologie du développement et de la croissance
Parmi les régions soumises à empreinte, certaines sont plus particulièrement impliquées dans des pathologies pédiatriques. Par exemple la dérégulation de l’expression des gènes SNRPN, IPW, UBE3A dans la région 15 q11-13 est associée aux syndromes de Prader-Willi ou d’Angelman en fonction de l’anomalie touchant respectivement l’allèle d’origine paternelle ou maternelle. Le syndrome de Prader-Willi est causé par la perte d’expression du gène SNRPN paternel et le syndrome d’Angelman par l’absence de l’expression du gène UBE3A d’origine maternelle [5].
Concernant la région 11p15, certains gènes sont exprimés à partir de l’allèle paternel (IGF-2, KCNQ1OT1), et d’autres à partir de l’allèle maternel (H19 et CDKN1C) (Fig 2A). La perte de l’allèle d’origine maternelle, structurale dans 25 % (unidisomie paternelle, c’est-à-dire perte de l’allèle maternel et duplication de l’allèle paternel) ou fonctionnelle par anomalie épigénétique ou épimutation (déméthylation au locus KCNQ1OT1 sur l’allèle maternel dans 60 % des cas, ou hyperméthylation de la région ICR1/H19 sur l’allèle maternel dans 10 % des cas (Fig 2C) engendre un syndrome de croissance excessive : le syndrome de Beckwith-Wiedemann (BWS), avec une surexpression d’IGF-2 (facteur de croissance), de KCNQ1OT1 (ARN non codant), l’extinction de H19 (ARN non codant) et de CDKN1C (inhibiteur du cycle cellulaire) ; enfin dans 5 % des cas une mutation de ce dernier gène peut être impliquée. Le BWS s’accompagne d’une croissance fœtale (taille et poids de naissance) et post-natale excessive, d’une viscéromégalie, d’une macroglossie, d’anomalies du développement telles qu’omphalocèle, hernie ombilicale ou hypertrophie hémi-corporelle. Des tumeurs (néphroblastome ou tumeur de Wilms, neuroblastome, hépatoblastome, corticosurrénalome, rhabdomyosarcome) sont associées dans environ 10 % des cas, cette fréquence pouvant varier de 2 à 30 % des cas selon l’anomalie moléculaire en cause (centromérique ou télomérique) [6-7].
À l’inverse, une perte de méthylation de l’allèle paternel , strictement localisée au centre local régulateur d’empreinte (ICR1) de la région 11p15 régulant IGF-2 et H19 (Fig 2B), est la cause la plus fréquente du syndrome de Silver-Russell (RSS) par extinction d’IGF-2 et surexpression d’H19 [8-9]. Ce syndrome, miroir du syndrome du BWS, associe un retard de croissance intra-utérin et post-natal avec périmètre crânien normal, une asymétrie hémi-corporelle de type hypotrophie, des anomalies faciales caractéristiques et des troubles alimentaires parfois majeurs.
Aide médicale à la procréation et pathologies de l’empreinte
L’analyse de ces pathologies de la croissance fœtale, nous a permis de montrer la prévalence élevée des procédures d’assistance médicale à la procréation (AMP) dans la population BWS et dernièrement de RSS [8, 10-12]. L’anomalie toujours épigé- nétique et d’empreinte (déméthylation du gène maternel KCNQ1OT1/ICR2 ou déméthylation de l’allèle paternel d’ ICR1 respectivement dans le BWS et le RSS) suggère que l’AMP empêche le maintien des marques de méthylation des gènes maternels dans les jours suivants la fécondation. Des anomalies de nombreux autres loci soumis à l’empreinte parentale [11, 12] viennent d’être mises en évidence et dans les BWS et dans les RSS évoquant un facteur agissant en « trans » qui va dans le sens de l’hypothèse récemment émise d’un réseau liant entre eux les gènes soumis à l’empreinte [13].
Une étude prospective exhaustive d’anomalies de l’empreinte génomique chez des enfants issus d’AMP devrait permettre d’impliquer ou d’infirmer telle ou telle procédure d’AMP, de stimulation ovarienne ou de la cause d’hypofertilité dans l’origine de ces perturbations épigénétiques.
Certains arguments plaident en faveur de l’implication des procédures d’AMP comme l’aspect en mosaïque cellulaire des anomalies épigénétiques apparaissant donc en période post zygotique [5, 6]. Cependant les études préliminaires n’impliquent pas une procédure particulière : Fertilisation In Vitro, ICSI, milieu de culture, congélation d’embryon, délai du transfert. D’autres études mettent en cause la sur-stimulation ovarienne qui survient lors de la phase terminale de la mise en place de l’empreinte de l’oocyte et dans certains cas, la cause de l’hypofertilité est suspectée devant des anomalies d’empreinte présentes dans le sperme parental ;
mais ces nombreuses publications sont discordantes et nécessitent un complément de recherche [14-20].
Étiologies des anomalies de l’empreinte
Quel est (sont) le défaut moléculaire initial de ces anomalies d’empreinte de la région 11p15 ?
Ces anomalies semblent apparaître en période post fertilisation (que ce soit pour les enfants issus d’AMP ou non) et ceci repose sur les arguments tels que la distribution en mosaïque de l’épimutation (BWS et SRS) ou l’existence de jumeaux monozygotes discordants (BWS/Normal, SRS/Normal) [5-7]. Cependant ces anomalies pourraient aussi être induites durant la mise en place de l’empreinte pendant la gamétogénèse et n’apparaître qu’en post zygotique.
Enfin l’impact de l’environnement est également probable comme le suggère les anomalies associées à l’AMP, mais aussi dans les situations de troubles nutritionnels lors de la gestation [21-23] ou les différences épigénétiques survenant au cours de la vie des jumeaux monozygotiques [24].
Certaines anomalies génétiques de type mutations dans les centres d’empreinte ICR 1 et 2 ont été décrites et devront être recherchées de façon plus systématique (séquençage des sept sites de liaison de CTCF de ICR1 par exemple). Les conseils génétiques à donner aux familles seraient alors différents. Enfin ces anomalies épigénétiques peuvent être imputées à d’autres anomalies génétiques concernant des gènes régulateurs de l’empreinte tels que les dnmt3aet 3L (établissement de l’empreinte lors de la gamétogénèse) ou dnmt1 (maintien de l’empreinte lors de la vague de démé- thylation globale en période préimplantatoire, avant le stade blastocyte) ou d’autres nouveaux gènes candidats comme PGC7/Stella, ZFP57, NLRP2 ou Boris [12].
Très récemment, nous avons montré que deux facteurs de pluripotence (OCT4 et SOX2) se lient au centre d’empreinte ICR1 et que des mutations de leurs sites de liaison sur l’allèle d’origine maternelle abolissent leur liaison et résultent en un gain de méthylation [25]. Ces facteurs de pluripotence ont été très récemment impliqués dans une autre forme d’empreinte, à savoir l’inactivation du chromosome X [26].
Ces résultats suggèrent que les facteurs de pluripotence jouent un rôle dans l’acquisition différentielle de la méthylation de l’ADN. OCT4 et SOX2 sont présents dans l’ovocyte et transmis au zygote. Leurs fonctions en terme de protection contre la méthylation de l’ADN lors de la maturation ovocytaire doit maintenant être testée.
Les analyses moléculaires à haut débit devraient permettre dans le futur de mieux appréhender l’impact des causes génétiques ou environnementales de ces anomalies épigénétiques.
CONCLUSION
Ainsi la régulation de l’expression des gènes est sous la dépendance du code génétique mais aussi du code épigénétique. À côté des anomalies génétiques des zones promotrices et de leur régulation transcriptionnelle, celles touchant les mécanismes épigénétiques entraînent des anomalies de l’expression des gènes conduisant à diverses pathologies développementales et tumorales. Le nombre de pathologies pédiatriques impliquant des anomalies d’empreinte parentale seront probablement dévoilées dans les années futures et la connaissance de leurs mécanismes pathogéniques permettront le développement de nouveaux outils diagnostiques. Le développement des thérapeutiques épigénétiques modulant les activités enzymatiques (DNA Méthyl transférase, histone déacétylase…) est envisagé pour essayer de contrôler les modifications des histones et de l’ADN notamment dans un but antitumoral [27]. Cependant les thérapies épigénétiques comportent des risques potentiels, puisque les actions sont non spécifiques d’un gène, mais globales et peuvent engendrer notamment la réactivation d’oncogènes silencieux. Une compré- hension plus approfondie de ces mécanismes épigénétiques est nécessaire pour aboutir à une meilleure utilisation de ces thérapeutiques.
REMERCIEMENTS
Les financements des recherches ont été accordés par l’INSERM, l’UPMC/Paris 6, le Programme National de Recherche en Reproduction et en Endocrinologie de l’Inserm, l’Agence de Biomédecine, le Ministère National de l’Enseignement, de la Recherche et de la Technologie, l’ANR.
BIBLIOGRAPHIE [1] Reik W., Dean W., Walter J. — Epigenetic reprogramming in mammalian development.
Science , 2001, 293 , 1089-93.
[2] McGrath J., Solter D. — Completion of embryogenesis requires both the maternal and paternal genomes. Cell , 1984, 37, 179-83.
[3] Dechiara T.M., Robertson E.J., Efstratiadis A. — Parental imprinting of the mouse insulin-like growth factor II gene. Cell , 1991 Feb 22, 64 , 849-59.
[4] Le Bouc Y., Gicquel C., Holzenberger M. — Physiology of somatotropic axis: interest of gene inactivation experiments. Bull. Acad. Natl. Med ., 2003, 187(7), 1225-43, discussion 1244-7.
[5] Nicholls R.D., Saitoh S., Horsthemke B. — Imprinting in Prader-Willi and Angelman syndromes. Trends Genet , 1998, 14 , 194-200.
[6] Schneid H., Seurin D., Vazquez M.P., Gourmelen M., Cabrol S., Le Bouc Y. — Parental allele-specific methylation of the human insulin — like growth factor II gene and WiedemannBeckwith syndrome. J. Med. Genet ., 1993, 30 , 353-362.
[7] Gaston V., Le Bouc Y., Soupre V., Burglen L., Donadieu J., Oro H., Aubry G., Vazquez M-P., Gicquel C. — Analysis of the methylation status of the KCNQ10T and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedmann syndrome. Eur. J. of Hum. Genet ., 2001, 9 , 409-418.
[8] Gicquel C., Rossignol S., Cabrol S., Houang M., Steunou V., Barbu V., Danton F., Thibaud N, Merrer ML, Burglen L, Bertrand AM, Netchine I, Le Bouc Y. — Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat. Genet ., 2005, 37 , 1003-1007.
[9] Netchine I., Rossignol S., Dufourg M.N., Azzi S., Rousseau A., Perin L., Houang M., Steunou V., Esteva B., Thibaud N., Raux Demay M.C., Danton F., Petriczko E., Bertrand A.M., Heinrichs C., Carel J.C., Loeuille G.A., Pinto G., Jacquemon M.L., Gicquel C., Cabrol S., Le Bouc Y. — 11p15 ICR1 loss of methylation is a common and specific cause of typical Russell-Silver Syndrome: clinical scoring system and epigenetic-phenotypic correlations. J. Clin. Endocrinol. Metab ., 2007, 92 , 3148-154.
[10] Gicquel C., Gaston V., Mandelbaum J., Siffroi J.P., Flahaut A., Le Bouc Y. — In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am. J. Hum. Genet ., 2003, 72, 1338-1341.
[11] Rossignol S., Steunou V., Chalas C., Kerjean A., Rigolet M., Viegas-Pequignot E., Jouannet P., Le Bouc Y., Gicquel C. — The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J. Med. Genet ., 2006, 43 , 902-907.
[12] Azzi S., Rossignol S., Steunou V., Sas T., Thibaud N., Danton F., Le Jule M., Heinrichs C., Cabrol S., Gicquel C., Le Bouc Y., Netchine I. — Multilocus methylation analysis in a large cohort of 11p15-related fœtal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci.
Hum. Mol. Genet ., 2009, Dec. 15, 18(24), 4724-33. Epub 2009 Sep 14.
[13] Gabory A., Ripoche M., Le Digarcher A., Watrin F., Ziyyat A., Forné T., Jammes H., Ainscough J.F.X, Surani A., Journot L., Dandolo L. — H19 acts as a trans regulator of the imprinted gene network controlling growth in mice Development 136 , 3413-3421 (2009) doi:10.1242/dev.036061.
[14] Market-Velker B.A., Zhang L., Magri L.S., Bonvissuto A.C., Mann M.R. — Dual effects of superovulation: loss of maternal and paternal imprinted methylation in a dose-dependent manner. Hum. Mol. Genet ., 2010 Jan 1, 19(1) , 36-51.
[15] Iura H., Obata Y., Komiyama J., Shirai M., Kono T. — Oocyte growth-dependent progression of maternal imprinting in mice. Genes Cells . 2006 Apr.11(4), 353-61.
[16] Sato A., Otsu E., Negishi H., Utsunomiya T., Arima T. — Aberrant DNA methylation of imprintedlociinsuperovulatedoocytes. Hum.Reprod .,2007Jan., 22(1) ,26-35.Epub2006Aug21.
[17] Tierling S., Souren N.Y., Gries J., Lo Porto C., Groth M., Lutsik P., Neitzel H., Utz-Billing I., Gillessen-Kaesbach G., Kentenich H., Griesinger G., Sperling K.,
Schwinger E., Walter J. — Assisted reproductive technologies do not enhance the variability of DNA methylation imprints in human. J. Med Genet . 2009 Nov 30. [Epub ahead of print].
[18] Fauque P., Jouannet P., Lesaffre C., Ripoche M.A., Dandolo L., Vaiman D, Jammes H. — Assisted Reproductive Technology affects developmental kinetics, H19 Imprinting Control Region methylation and H19 gene expression in individual mouse embryos. BMC Dev Biol ., 2007 Oct. 18, 7 , 116.
[19] Kobayashi H., Sato A., Otsu E., Hiura H., Tomatsu C., Utsunomiya T., Sasaki H., Yaegashi N., Arima T. — Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum. Mol. Genet ., 2007 Nov. 1, 16 (21) , 2542-51. Epub 2007 Jul 17.
[20] Boissonnas C.C., Abdalaoui H.E., Haelewyn V., Fauque P., Dupont J.M., Gut I., Vaiman D., Jouannet P., Tost J., Jammes H. — Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile men. Eur. J. Hum. Genet ., 2010, Jan., 18 (1), 73-80. Epub.
[21] Heijmans B.T., Tobi E.W., Stein A.D., Putter H., Blauw G.J., Susser E.S., Slagboom P.E., Lumey L.H. — Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. U S A , 2008 Nov. 4, 105 (44), 17046-9. Epub 2008 Oct. 27.
[22] McMillen I.C., Robinson J.S. — Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol. Rev ., 2005 Apr., 85 (2), 571-633. Review.
[23] Gicquel C., El-Osta A., Le Bouc Y. — Epigenetic regulation and fetal programming.
Best
Pract. Res. Clin. Endocrinol. Metab ., 2008 Feb., 22 (1) , 1-16.
[24] Fraga M.F., Ballestar E., Paz M.F., Ropero S., Setien F., Ballestar M.L., Heine-Sun˜er D., Cigudosa J.C., Urioste M., Benitez J., Boix-Chornet M., Sanchez-Aguillera A., Ling C., Carlsson E., Poulsen P., Vaag A., Stephan Z., Spector T.D., Wu Y.Z., Plass C., Esteller M. — Epigenetic differences arise during the lifetime of monozygotic twins. Proc.
Natl. Acad. Sci. U S A , 2005, Jul. 26, 102 (30), 10604-9. Epub 2005 Jul. 11.
[25] Demars J., Shmela ME., Rossignol S., Okabe J., Netchine I., Azzi S., Cabrol S., Le Caignec C., David A., Le Bouc Y., El-Osta A., Gicquel C. — Analyses of the IGF2/H19 imprinting control region uncovers nex genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. How Mol Genet. 2010 Mar 1 ; 19 (5) :
803-14. Epub 2009 Dec. 9.
[26] Donohoe ME, Silva SS, Pinter SF, Xu N, Lee JT. 8 The pluripotency factor Oct 4 interacts with Ctcf and also controls X-chromosome pairing and counting. Nature. 2009 Jul 2 ; 460 (7251) : 128-32. Epub 2009 Jun 17.
[27] Egger G., Liang G., Aparicio A., Jones P.A. — Epigenetics in human disease and prospects for epigenetic therapy. Nature , 2004, 429 , 457-463.
DISCUSSION
M. Yves VILLE
Certaines études rapportent, outre l’augmentation de fréquence de syndromes soumis à empreinte, une aggravation du phénotype attendu après PMA. Retrouve-t-on une corrélation entre le degré de méthylation différentielle et la sévérité du phénotype 3 ?
Il existe effectivement une augmentation de fréquence des cas de syndrome de Beckwith Wiedemann dû à des anomalies d’empreinte chez des enfants issus d’AMP, mais je ne pense pas qu’on puisse dire qu’il s’agisse d’un phénotype plus grave dans ces situations et au contraire ce n’est pas le cas pour le syndrome de Beckwith Wiedemann puisque dans les cas issus d’AMP l’anomalie moléculaire n’est pas associée à un risque de tumeur embryonnaire. L’hypothèse d’une corrélation entre le degré de l’anomalie de méthylation et la sévérité du phénotype est tout à fait justifiée, cependant comme nos études portent le plus souvent sur un seul tissu, les globules blancs circulants, et qu’il existe une très grande variabilité inter-tissulaire du pourcentage de cellules présentant l’anomalie épigénétique par rapport aux cellules normales (mosaïque tissulaire), il est difficile d’établir cette corrélation de façon certaine.
M. Roger NORDMANN
De nombreux travaux récents ont démontré que les performances scolaires des adolescents issus de mères ayant consommé de l’alcool pendant leur grossesse sont notablement inférieures à celles d’un groupe témoin. L’alcool représente ainsi la cause majeure de retard mental d’origine non génétique. Dispose-t-on de données permettant de définir dans leur ensemble les mécanismes des altérations épigénétiques liées à l’alcoolisation pendant la grossesse ? Certaines publications indiquent qu’un retard de développement affecte l’enfant lorsque le père est en difficulté avec l’alcool alors même qu’il n’y a pas eu d’alcoolisation maternelle pendant la grossesse.Uneempreintepaternelleest-elledocumentéedanscesconditions ?
Il existe effectivement quelques travaux montrant que l’alcool, comme d’ailleurs le tabac, induit des modifications de la méthylation de l’ADN ainsi que des modifications de l’acétylation des histones. Ceci peut être rapproché de ce que l’on observe dans d’autres situations environnementales comme les troubles nutritionnels, le stress. Je ne me suis pas intéressé particulièrement à l’impact de l’alcool dans ces anomalies, mais effectivement il n’est pas impossible que l’alcool lors de la gestation soit néfaste via des modifications épigénétiques. Concernant la responsabilité du père, il existe quelques travaux montrant par exemple qu’une altération génétique d’enzymes impliqué dans la méthylation de l’ADN et entraînant donc une anomalie épigénétique lors de la spermatogénèse soit à l’origine de pathologie chez le fœtus et l’enfant. On peut donc imaginer qu’une alcoolisation chronique paternelle puisse entraîner des anomalies épigénétiques directement lors de la gamétogénèse et soit responsable de pathologies, alors que la mère est indemne.
M. Yves LE GALL
Vous avez dit que les phénomènes épigénétiques « n’étaient pas contenus dans la séquence de l’ADN ». Ils sont pourtant le fait de systèmes enzymatiques codés par la séquence nucléotidique et qui intervient suivant un programme déterminé au cours de la différentiation cellulaire.
Oui bien sûr les enzymes qui régulent l’état de méthylation de l’ADN ou de l’acétylation des queues d’histones sont codées par des gènes et donc dépendent de la séquence nucléotidique, mais l’épigénétique est définie comme les modifications d’un segment de la chromatine (qu’elles soient méthylation, acétylation, changement de conformation chromatinienne : ouverte ou compacte) et dont les régulateurs ne sont pas contenues dans la séquence même d’ADN de cette portion de chromatine, ces modifications persistent, sont transmissibles de façon mitotique et contrôlent l’expression des gènes autour de cette partie de chromatine.
M. Claude JAFFIOL
Les troubles métaboliques (obésité-diabète) apparus chez des enfants nés de mères présentant les mêmes troubles en période préconceptionnelle correspondent-ils à des modifications épigénétiques ? Ces modifications sont-elles susceptibles d’une transmission sur plusieurs générations ?
Beaucoup de travaux concernent actuellement l’impact des troubles nutritionnels et métaboliques durant la grossesse, en période néonatale précoce et effectivement aussi dans la période pré-conceptionnelle, notamment lors des cycles précédant l’ovulation et la conception. Dans ce sens, l’étude récente de la méthylation de la région où est situé le gène d’IGF 2 chez les personnes nées lors de la famine de l’hivers 1944-1945 en Hollande montre bien l’impact de la nutrition dans la première partie de la grossesse, impact persistant même chez ces personnes âgées de plus de 60 ans. Les études réalisées chez les animaux montrent qu’il peut y avoir une transmission sur quelques générations mais qu’en une ou deux générations cela s’estompe si l’élément déclencheur (par exemple dénutrition lors de la gestation) n’existe plus.
M. Georges DAVID
Vous avez terminé sur le mécanisme épigénétique en cause dans le « syndrome de Barker », c’est-à-dire dans les risques à long terme de l’hypotrophie fœtale. Par ailleurs, on constate après fécondation in vitro , dans des grossesses uniques, une hypotrophie fœtale, celle-ci pourrait-elle être le maillon d’un enchaînement conduisant aux pathologies de l’adulte précédemment citées ?
De façon générale ces atteintes lors de la période fœtale quelles qu’en soient les causes :
hypovascularisation placentaire, corticoïdes, stress, dénutrition ou autre cause environnementale peuvent provoquer des modifications épigénétiques permanentes entraînant en fonction de la vie postnatale rencontrée (par exemple surnutrition) des pathologies retardées de type résistance à l’insuline, obésité, pathologies cardiovasculaires ou inadaptation au stress.
M. Jacques ROCHEMAURE
Le syndrome de Wiedemann Beckwith peut-il être transmissible et avec quelle fréquence ?
C’est une question importante qui est souvent posée aux généticiens. Bien sûr quand il s’agit d’un cas avec mutation de CDKN1C (inhibiteur des cyclines-cdk de la phase G1 du cycle cellulaire) la transmission par la mère va obligatoirement entraîner la pathologie, par contre le plus souvent le conseil génétique est difficile. La fréquence globale est de 1/13700 et on ne sait pas prédire pour l’instant si une anomalie de méthylation est due à un phénomène exceptionnel ou s’il est induit par une anomalie d’un gène codant pour une enzyme de type « DNA methyl transferase », ou d’une mutation ou délétion dans des régions de type centre d’empreinte. Le conseil génétique est de même difficile dans les cas d’isodisomie paternelle.
M. Pierre GODEAU
Des mécanismes immunologiques peuvent-ils s’opposer aux mécanismes épigénétiques normaux ? C’est-à-dire lorsqu’il y a des anticorps anti ADN et/ou antihistones, comme dans nombre de maladies autoimmunes. Peuvent-ils s’opposer et/ou modifier la méthylation de l’ADN ?
C’est un domaine en dehors de ma compétence mais votre question est très pertinente et intéressante méritant que des études soient réalisées dans ce sens. Le lupus érythémateux est associé à des anticorps anti-H1, et anti-H3, mais je ne sais pas si on a analysé leurs répercussions au niveau de la méthylation de l’ADN ou de la modification des histones.
M. André VACHERON
Pour les parents, quel est le risque d’avoir un second enfant atteint du syndrome de Wiedemann Beckwith dans une fratrie, après un premier cas ?
S’il est facile de répondre dans le cas d’un premier enfant atteint d’une mutation du gène CDKN1C transmis par la mère et engendrant un risque de 100 %, il est impossible dans l’état de nos connaissances actuelles d’aller plus loin dans le conseil génétique apporté aux parents pour les autres anomalies épigénétiques, sauf si une mutation/délétion a été auparavant mise en évidence par exemple dans un centre d’empreinte (situation actuellement rarement rencontrée).
M. Paul VERT
Les travaux présentés font évoquer des notions anciennes que l’on avait pensé obsolètes :
l’héridité des caractères acquis, l’hérédo-alcoolisme. Faut-il revisiter ces notions que les lois de Mendel avaient fait écarter ?
Il est sûr que des modifications épigénétiques acquises lors d’un environnement particulier peuvent se transmettre et continuer au moins sur une ou deux générations aprés disparition des facteurs inducteurs de ces anomalies. Ces résultats ont été obtenus lors d’expérimentations animales par des modifications des apports nutritionnels. L’hypothèse est qu’il existerait ainsi une adaptation rapide à un changement environnemental (ici par exemple à un environnement pauvre en ressources énergétiques induisant un petite taille plus adaptée à ces restrictions en post natal). Certains études épidémiologiques et moléculaires chez l’homme sont actuellement réalisées et permettront ultérieurement de conclure pour l‘espèce humaine et notamment sur la transmission au moins temporaire de facteurs acquis après différentes situations : nutritionnelles, stress maternel etc. Ceci ouvre bien sur des changements de concepts.
M. Jean-Pierre NICOLAS
L’importance des méthylations n’est-elle pas bien démontrée par les malformations (spina bifida, non fermeture de la gouttière neurale, etc.) observées dans les carences en folates (de donneurs de groupements méthyle) chez les femmes gestantes.
Je ne suis pas un spécialiste de la spina bifida mais il est possible qu’elle puisse résulter comme d’autres malformations du système nerveux d’anomalies épigénétiques de diffé- rents facteurs impliqués en épigéntique tels que Zac1 ou Suz12. Vous avez tout à fait raison concernant les carences en folates et les relations avec certaines pathologies.
Effectivement de nombreuses expériences ont montré que durant certaines périodes ontogéniques critiques, mais aussi en post natal, la méthylation de l’ADN est très dépendante de la disponibilité en donneurs de méthyl et des cofacteurs incluant, méthionine, choline, acide folique et Vit B12 apportées par la nutrition. Un autre modèle du contrôle par la nutrition des modifications épigénétiques est apporté par le modèle de la souris Agouti (Avy). Le gène agouti est impliqué dans la production d’un pigment jaune dans les mélanocytes. L’allèle agouti Avy résulte d’une insertion d’un transposon en amont et induit une expression ectopique du pigment jaune associée à une obésité à l’état adulte et ceci quand le transposon est dans un état déméthylé. La supplémentation maternelle en méthyl avec des cofacteurs tels que la vitamine B12 ou des folates augmentent la méthylation de l’ADN du transposon résultant en un pelage brun et une protection contre l’obésité.
Bull. Acad. Natle Méd., 2010, 194, no 2, 287-300, séance du 16 février 2010